
6-溴咪唑并[1,2-b]哒嗪-3-甲酸的合成
2021-09-01 12:46张向龙赵雷振范亚锋油天钰袁文鹏
合成化学 2021年8期
程 伟, 张向龙, 赵雷振, 范亚锋, 油天钰, 袁文鹏*
(1. 齐鲁工业大学(山东省科学院) 山东省科学院菏泽分院 山东省生物工程技术创新中心,山东 菏泽 274000; 2. 山东友帮生化科技有限公司 山东省小分子杂环化合物工程实验室,山东 菏泽 274000)
哒嗪类化合物是一类重要的杂环化合物,在医药[1-3]、农药[4-5]、光电材料[6-7]等领域得到了大量使用,具有较高的应用价值。咪唑并[1,2-b]哒嗪衍生物是哒嗪类化合物中生物活性较强的一类,广泛应用于抗菌药物[8-9]、抗炎药物[10]、抗寄生虫药物[11-12]、抗癌药物[13-15]以及治疗帕金森疾病药物[16-17]的研发与生产。
6-卤代咪唑并[1,2-b]哒嗪-3-甲酸是一种重要的抗癌新药中间体,在白细胞介素-1受体相关激酶4(IRAK4)抑制剂[13]、原肌球蛋白受体激酶(TRK)抑制剂[18-21]、异源二聚细胞因子(IL-2)抑制剂[22]、蛋白激酶(PRK)调节剂[23]、溴结构域蛋白(CBP/EP300)抑制剂[24]、间变性淋巴瘤激酶(ALK)抑制剂[25-26]等抗癌新药的开发工作中表现出优异的生物活性,已有多个相关药物进入临床二期、三期实验,肿瘤抑制效果显著,具有重要的临床应用价值[27]。
目前,文献报道的6-氯咪唑并[1,2-b]哒嗪-3-甲酸的合成方法主要有两种:Bi等[13]以6-氯咪唑并[1,2-b]哒嗪-3-甲醛为原料,经亚氯酸钠氧化过夜制备6-氯咪唑并[1,2-b]哒嗪-3-甲酸。该方法原材料价格昂贵,需要柱层析纯化,且单步收率只有67%,不适合工业化放大。Liu等[28]先用甲酸乙酯和氯乙酸乙酯在乙醇钠催化下反应3 d合成2-氯-2-甲酰乙酯,再与3-氨基-6-氯哒嗪反应制备6-氯咪唑并[1,2-b]哒嗪-3-甲酸乙酯,再经过氢氧化钠水解共3步制备6-氯咪唑并[1,2-b]哒嗪-3-甲酸。该方法反应时间长,中间体2-氯-2-甲酰乙酯性质不稳定且收率低,反应过程操作复杂不易放大生产,且总收率不到40%,难以大规模推广。6-溴咪唑并[1,2-b]哒嗪-3-甲酸的合成方法目前尚无文献报道,具有较高的研究价值。
本文提供了一种6-溴咪唑并[1,2-b]哒嗪-3-甲酸的合成新方法,以水合肼和马来酸酐为起始原料,经缩合、溴化、氨解、亚胺化、关环缩合、水解反应共6步反应得到目标产物(Scheme 1)。该方法具有原料易得、成本低廉、操作简单和收率较高等优点。
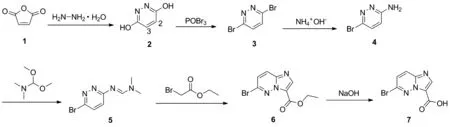
Scheme 1
1 实验部分
1.1 仪器与试剂
YRT-3型熔点仪;Bruker AV-III-400 MHz型核磁共振仪(DMSO-d6为溶剂,TMS 为内标);Avatar 370型红外光谱仪(KBr压片);Vario EL型元素分析仪;LC-16型液相色谱仪测定。
所用试剂均为分析纯,国药集团化学试剂有限公司。
1.2 合成
(1) 3,6-二羟基哒嗪(2)的合成
将水合肼145 g(2.32 mol)和1 L水加入到反应瓶中,控温不高于10 ℃,滴加浓盐酸228 g(2.32 mol),滴毕,分四批加入顺丁烯二酸酐(1)196 g(2.00 mol),升温至回流,反应4 h。降温至室温,有大量固体析出,抽滤,滤饼用200 mL水洗涤,于55 ℃真空干燥得215 g化合物2,白色固体,收率95.9%,纯度99.2%[HPLC 归一化法:色谱柱为Agilent Eclipse Plus C18 柱(4.6 mm×250 mm,5 μm),流动相A为纯化水,流动相B为甲醇,梯度洗脱条件:0~2 min, A: 60%; 2~8 min, A: 50%; 8~20 min, A: 30%; 检测波长254 nm,柱温30 ℃,流速1.0 mL/min,进样量20 μL,下同]; m.p.298~300 ℃;1H NMR(DMSO-d6, 400 MHz)δ: 7.01(s, 2H, H-2, H-3), 11.56(s, 2H, -OH) ;13C NMR (DMSO-d6, 100 MHz)δ: 130.8, 156.8; IRν: 3086, 2963, 1660, 1552, 1411, 1275, 1011, 899, 845, 813 cm-1; Anal. calcd for C4H4N2O2: C 42.86, H 3.60, N 24.99, found C 42.82, H 3.69, N 24.93。
(2) 3,6-二溴哒嗪(3)的合成
称取化合物2112 g(1.00 mol)加入1.5 L甲苯中,搅拌使其混合均匀;分三批加入三溴氧磷630 g(2.20 mol),升温至100 ℃,反应5 h。将反应液缓慢倒入3 L冰水中,充分搅拌,控温不高于10 ℃,用碳酸氢钠调节pH至中性,用二氯甲烷(2×500 mL)萃取,合并有机相,依次用水(2×300 mL)和饱和食盐水(2×200 mL)洗涤,减压蒸除溶剂得粗品,用正己烷重结晶得淡黄色固体3195 g,收率82.2%,纯度98.7%; m.p.115~116 ℃;1H NMR(DMSO-d6, 400 MHz)δ: 8.01(s, 2H, H-2, H-3);13C NMR(DMSO-d6, 100 MHz)δ: 134.9, 148.4; IRν: 3414, 3093, 1545, 1387, 1157, 1110, 852, 754 cm-1; Anal. calcd for C4H2N2Br2: C 20.20, H 0.85, N 11.78, found C 20.23, H 0.87, N 11.81。
(3) 3-氨基-6-溴哒嗪(4)的合成
将化合物3464 g(1.95 mol)加入装有氨水(25%)1.05 L(13.62 mol)的3 L高压釜中,通氮气置换空气两次,氮气充压1.0 MPa,搅拌并升温至100 ℃,反应7 h。于室温搅拌1 h,过滤,滤饼用水(2×200 mL)洗涤,于55 ℃真空干燥得淡黄色固体4290 g,收率85.7%,纯度98.2%; m.p.192~194 ℃;1HNMR(DMSO-d6, 400 MHz)δ: 6.63(s, 2H, -NH2), 6.76(s, 1H, H-2), 7.42(s, 1H, H-3);13C NMR(DMSO-d6, 100 MHz)δ: 118.0, 132.4, 136.0, 160.8; IRν: 3152, 1641, 1593, 1452, 1146, 1051, 834, 647 cm-1; Anal. calcd for C4H4N3Br: C 27.61, H 2.32, N 24.15, found C 27.58, H 2.35, N 24.21。
(4)N′-(6-溴哒嗪-3-基)-N,N-二甲基甲脒(5)的合成
将化合物450 g(0.29 mol)分两次加入到N,N-二甲基甲酰胺二甲基缩醛350 g(2.89 mol)中,升温至90 ℃,反应5 h。减压浓缩至无液体流出,残余物加入150 mL甲醇,降温至-7 ℃搅拌2 h,有大量固体析出,抽滤,滤饼于-7 ℃用冻甲醇(2×50 mL)淋洗,烘干得白色固体560.2 g,收率91.3%,纯度98.4%; m.p.115~117 ℃;1H NMR(DMSO-d6, 400 MHz)δ: 3.01(s, 3H, -CH3), 3.13(s, 3H, -CH3), 7.05(s, 1H, H-2), 7.66(s, 1H, H-3), 8.21(s, 1H, -N=CH-);13C NMR(DMSO-d6, 100 MHz)δ: 34.8, 40.7, 125.8, 132.9, 141.4, 155.9, 163.9; IRν: 3068, 2909, 1625, 1562, 1383, 1114, 1062, 983, 858 cm-1; Anal. calcd for C7H9N4Br: C 36.70, H 3.96, N 24.46, found C 36.77, H 3.92, N 24.51。
(5) 6-溴咪唑并[1,2-b]哒嗪-3-甲酸乙酯(6)的合成
称取化合物550 g(0.22 mol)加入到装有500 mL DMF的1 L反应瓶中,滴加溴乙酸乙酯54.6 g(0.33 mol),滴毕,加入碘化钾1.8 g(11 mmol),升温至90 ℃,反应8 h,降温至室温,加入1.5 L水搅拌20 min,有大量固体析出,抽滤,滤饼用水(2×200 mL)洗涤,乙酸乙酯重结晶得淡黄色固体652.1 g,收率87.7%,纯度97.9%; m.p.74~76 ℃;1H NMR(DMSO-d6, 400 MHz)δ: 1.28(t,J=7.1 Hz, 3H, -CH3), 4.31(q,J=7.2 Hz, 2H, -CH2-), 8.01(d,J=9.6 Hz, 1H, H-3), 8.10(d,J=9.6 Hz, 1H, H-2), 8.67(s, 1H, H-8);13CNMR(DMSO-d6, 100 MHz)δ: 13.5, 63.5, 125.8, 126.5, 136.2, 139.4, 158.1, 159.1, 168.0; IRν: 3105, 2978, 1723, 1514, 1435, 1343, 1265, 1188, 1048, 822, 760 cm-1; Anal. calcd for C9H8N3O2Br: C 40.02, H 2.99, N 15.56, found C 39.97, H 3.02, N 15.53。
(6) 6-溴咪唑并[1,2-b]哒嗪-3-甲酸(7)的合成
将化合物635 g(0.13 mol)加入到200 mL水中,搅拌使其混合均匀;加入氢氧化钠13 g(0.33 mol),反应8 h。用10 %稀盐酸调pH至中性,有大量固体析出,抽滤,滤饼用水(2×200 mL)洗涤,于55 ℃真空干燥得类白色固体730.2 g,收率96.1%,纯度99.1%; m.p.215~217 ℃;1H NMR(DMSO-d6, 400 MHz)δ: 7.64(d,J=9.6 Hz, 1H, H-3), 8.22(d,J=9.5 Hz, 1H, H-2), 8.31(s, 1H, H-8), 13.01(s, 1H, -COOH);13C NMR(DMSO-d6, 100 MHz)δ:120.7, 125.0, 128.2, 138.5, 140.8, 141.2, 159.5; IRν: 3433, 3097, 1705, 1528, 1438, 1358, 1274, 1161, 1105, 832, 764 cm-1; Anal. calcd for C7H4N3O2Br: C 34.74, H 1.67, N 17.36, found C 34.77, H 1.70, N 17.31。
2 结果与讨论
2.1 化合物3的合成
化合物3的合成为溴化反应,选择三溴氧磷为溴化试剂,研究发现溴化试剂三溴氧磷的用量对实验结果影响较大。在确定反应温度100 ℃反应5 h的条件下,考察了三溴氧磷的用量对化合物3收率的影响,反应结果见表1。由表1中可以看出,三溴氧磷用量为化合物2的1.1倍时,收率达到最高。继续增加三溴氧磷的用量,会造成产物的分解,目标产物收率降低,所以选择n(POBr3)/n(化合物2)=1.1/1时为最优比例。

表1 三溴氧磷用量对收率和纯度的影响
2.2 化合物4的合成
化合物4的合成为氨解反应,本实验选取25%的氨水作为氨解试剂,该反应具有一定的选择性,氨水的用量和反应压力是该反应的主要影响因素。在确定氮气充压1.0 MPa,反应温度100 ℃反应7 h的条件下,考察氨水与原料的投料比对化合物4的收率影响,实验结果见表2。由表2可以看出,氨水量较少时反应不容易发生,表现为产品产率低;随着氨水的用量增加,产率不断提高。当氨水的用量为化合物3的3.5倍时,反应产率达到为85.7%,再增加氨水的用量,反应产率增长幅度不大,且容易造成物料浪费,因此选择原料摩尔配比为:n(氨水)/n(化合物3)=3.5/1。

表2 氨水用量对收率和纯度的影响
在实验研究中发现,加大压力有利于氨解反应的进行。在确定反应温度100 ℃反应7 h的条件下,考察了不同的反应压力对化合物4收率的影响,结果见表3。

表3 反应压力对收率影响
由表3可以看出,化合物4的收率随着压力的上升而不断增加。当压力为1.0 MPa时,反应收率最高,压力高于1.0 MPa时反应收率上升幅度不大,并随着反应压力增加对反应设备要求也会越高,反应操作难度加大,因此选择反应压力为1.0 MPa。
2.3 化合物6的合成
合成化合物6的反应为关环缩合反应,选用溴乙酸乙酯作为缩合试剂,碘化钾作为催化剂。反应过程通过结晶纯化高收率制得化合物6,避免了柱层析纯化,有利于工业化生产。本实验在确定反应温度和反应时间的情况下,考察了溴乙酸乙酯用量对化合物6反应收率的影响,结果见表4。
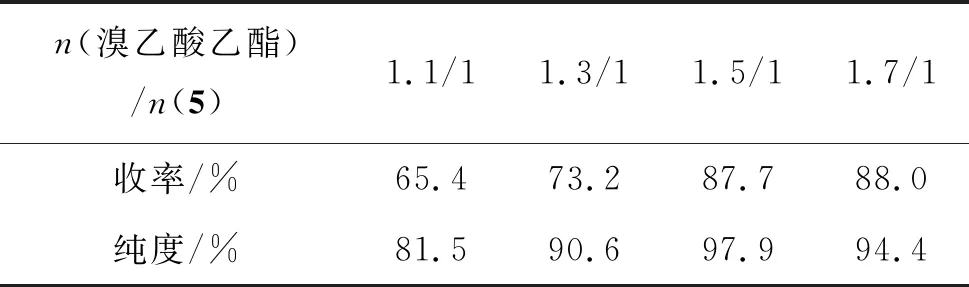
表4 溴乙酸乙酯用量对收率和纯度影响
由表4可以看出,溴乙酸乙酯的用量较少时反应不容易发生,表现为产品收率低;随着溴乙酸乙酯的用量增加,收率不断提高。当溴乙酸乙酯的用量为化合物5的1.5倍时,反应收率达到为87.7%;再增加溴乙酸乙酯的用量,反应收率增长幅度不大,且反应体系颜色变深,得到的产品结晶发粘,颜色较深,因此选择溴乙酸乙酯用量为:n(溴乙酸乙酯)/n(5)=1.5/1。
本实验还考察了结晶溶剂对反应收率的影响,实验结果见表5。由表5可以看出,以乙酸乙酯作为结晶溶剂,化合物6的收率最高,并且产品颜色为淡黄色固体。以二氯甲烷为结晶溶剂,产品母液损失较多,造成收率下降;用乙醇作溶剂时,析出固体发粘,颜色为褐色,收率也低。用正己烷作为结晶溶剂,需要溶剂量大,产品收率和纯度都较低。综上,选用乙酸乙酯为化合物6的结晶溶剂。
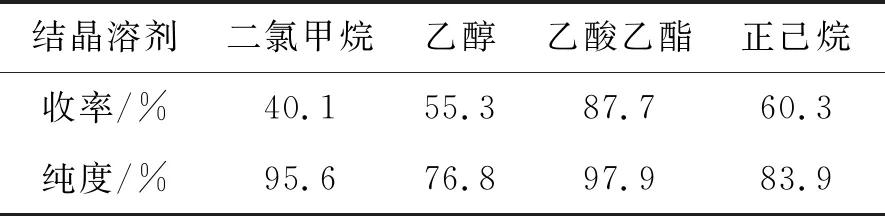
表5 结晶溶剂对收率和纯度影响
报道了一种6-溴咪唑并[1,2-b]哒嗪-3-甲酸的合成新方法。以水合肼和马来酸酐为起始原料,在盐酸催化下发生缩合反应,再与三溴氧磷进行溴代,与氨水发生氨解反应,再与N,N-二甲基甲酰胺二甲基缩醛进行亚胺化反应,与溴乙酸乙酯缩合成环,最后发生碱水解共6步反应合成了目标产物6-溴咪唑并[1,2-b]哒嗪-3-甲酸,总收率52.0%。该合成方法路线新颖、成本低廉、操作简单、收率高、中间反应过程采用结晶纯化,更适合工业化放大生产。
猜你喜欢
核化学与放射化学(2022年2期)2022-04-28
中学生理科应试(2021年10期)2021-12-07
中华养生保健(2020年3期)2020-11-16
饲料博览(2020年7期)2020-08-18
农药科学与管理(2019年9期)2019-11-23
科技视界(2016年26期)2016-12-17
中小企业管理与科技·中旬刊(2014年7期)2014-09-24
食品工业科技(2014年6期)2014-05-10
祝您健康(1985年3期)1985-12-30
中国青年(1965年18期)1965-08-20
