
吹扫捕集-气相色谱-质谱法快速测定同序列地下水和土壤中的37种挥发性有机物
2021-08-29 14:17张利钧杨丰春
理化检验-化学分册 2021年8期
付 瑶,张利钧,杨丰春,李 欣,任 伟
(济南市环境研究院,济南 250100)
挥发性有机物(VOCs)主要包括芳烃、卤代芳烃、卤代烷烃、烯烃类等,因其沸点低、分布广和“三致”(致癌、致畸、致突变)作用,严重危害人类健康和生态环境[1],被列为优先控制污染物。环境中的土壤和地下水关系紧密、相互作用,在实际工作过程中常会出现同时测定两者中相同指标的情况,且土壤和地下水污染均具有过程缓慢、不易发现、隐蔽性强和难以修复等特点,因此及时检测进而加强监管修复尤为重要。
目前检测土壤和地下水中VOCs含量的前处理方法主要包括吹扫捕集法[2-10]、顶空法[11-13]和顶空-固相萃取法[14]等。吹扫捕集作为一种动态顶空技术,可减少传统顶空法平衡溶剂带来的二次污染。气相色谱法常用的检测器有氢火焰离子化检测器(FID)[7,12-13]、光离子化检测器(PID)[15]。FID 无法定性区分色谱峰重叠时VOCs的种类,或是在基体组分复杂时呈现假阳性;PID 仅针对VOCs总量进行测定,且易受土壤含水率和空气的影响[15],两者均具有一定的局限性。气相色谱仪与质谱仪[4-8,13-14]联用,可快速、灵敏地对VOCs各组分进行定性分析和定量测定。
目前文献报道主要针对单一地下水或土壤的测试条件进行研究,而对于校准曲线的绘制、吹扫捕集条件优化等试验过程中常遇到的细节问题较少涉及。国内现行标准HJ 605-2011«土壤和沉积物挥发性有机物的测定 吹扫捕集/气相色谱-质谱法»及HJ 639-2012«水质 挥发性有机物的测定 吹扫捕集/气相色谱-质谱法»提供的吹扫捕集参数主要针对旧式冷阱,与应用于当前新式冷阱的参数相比有很大差异。
本工作根据相关质量标准要求[16-17],分别优化了地下水和土壤的校准曲线的绘制、吹扫捕集和气相色谱-质谱(GC-MS)条件,并对济南市7个水源地的土壤及地下水样品进行分析,以期为地下水和土壤的联合修复提供科学依据。
1 试验部分
1.1 仪器与试剂
岛津QP2020NX 型气相色谱-质谱联用仪;Tekmar ATOMX-XYZ型全自动固液一体吹扫捕集装置;MS205DU 型电子天平;CNW 40 mL 棕色吹扫瓶。
37种VOCs混合标准溶液:2.0 g·L-1,使用前用甲醇稀释至20 mg·L-1,于-20 ℃保存。
内标溶液:2.0 g·L-1,含氟苯、4-溴氟苯、1,2-二氯苯-d4等3种组分,于-20 ℃保存。
替代物标准溶液:250 mg·L-1,含二溴氟甲烷、甲苯-d8两种组分,于-20 ℃保存。
抗坏血酸为优级纯;甲醇为农残级;石英砂为分析纯;试验用水为超纯水(电导率18.2 MΩ·cm)。
1.2 仪器工作条件
1)吹扫捕集条件 吹扫流量40 mL·min-1,吹扫温度20 ℃,吹扫时间11 min;干吹时间,测定土壤样品时需2 min,测定地下水样品时需0.5 min;预脱附温度245 ℃,脱附温度250 ℃,脱附时间1.7 min;烘烤温度280 ℃,烘烤时间8 min;传输线温度180 ℃。
2)色谱条件 SHIMADZU SH-Rxi-624Sil MS色谱柱(60 m×0.32 mm,0.18μm);进样口温度200℃;载气为氦气(纯度大于99.999%);分流进样,分流比30∶1;柱流量1.5 mL·min-1;进样量1.0 μL。柱升温程序:初始温度为38 ℃,保持1.8 min;以10 ℃·min-1速率升温至120 ℃;再以15 ℃·min-1速率升温至240 ℃,保持2 min。
3)质谱条件 电子轰击离子源(EI);电离能量70 e V;离子源温度200℃,传输线温度280℃;选择离子监测(SIM)模式。其他质谱参数见表1。
1.3 试验方法
1.3.1 样品的采集、保存及测定
水样采样前,在吹扫瓶内加入25 mg 抗坏血酸。采集样品时,应尽量避免或减少样品在空气中的暴露,并使水样在瓶口形成上弯月面,不留气泡,冷藏,于14 d内测定。土壤样品采样前,在棕色吹扫瓶中放入一个清洁的磁力搅拌棒,采样前后密封并称重(精确至0.01 g),采样质量约5 g,冷藏,于7 d内测定。
样品经冷藏后取出,水样满瓶,用微量进样针移出相应体积的水样后,加入40μg·L-1内标溶液40μL;土壤样品加入10.00 mL水及40μg·L-1内标溶液10.00μL。再分别加入一定体积的替代物标准溶液,经涡旋仪振荡混匀后放入样品架,按仪器工作条件进行测定。
1.3.2 绘制校准曲线的混合标准溶液系列的配制
将适量的37 种VOCs混合标准溶液、替代物标准溶液、内标溶液用水稀释,并定容至50 mL 容量瓶中,配制成目标物和替代物质量浓度为1,2,4,8,16,32,64,80μg·L-1,内标质量浓度为40μg·L-1的混合标准溶液系列。其中,土壤样品绘制校准曲线时,取10.00 mL 上述混合标准溶液系列至吹扫捕集样品瓶中(内置5 g石英砂空白,实际目标物的质量浓度为2,4,8,16,32,64,128,160μg·kg-1);水样品绘制校准曲线时,取满瓶(约40 mL)。进样时,仪器用气密性注射器自动吸取混合标准溶液5.0 mL,按仪器工作条件测定,内标法定量。
2 结果与讨论
2.1 校准曲线绘制条件的优化
绘制校准曲线是多组分易挥发目标物的难点,特别是对于低沸点化合物,操作步骤对相对响应因子(RRF,为目标物响应值与内标响应值的比值与内标质量浓度与目标物质量浓度比值的乘积)及相关系数(r)的影响较大(图1)。土壤样品的混合标准溶液系列的优化首先选取两种配制方法:1#在吹扫瓶内加入5.00 mL 水后,用微量进样针加入混合标准溶液;2#采用50 mL 玻璃容量瓶配制,移液管移取5.00 mL混合标准溶液至吹扫瓶中。方法1#减少了配制过程中的损失,目标物的RRF 明显增大,RRF的相
对标准偏差(RSD,n=8)大部分低于20%,但由于存在系统误差(混合标准溶液系列各点实际的质量浓度不同程度地大于理论质量浓度),部分目标物RRF的RSD(n=8)在20%左右,且校准曲线的相关系数偏低。方法2#校准曲线的相关系数不小于0.990 0,线性良好,但RRF 较低,导致其RSD(n=8)普遍不低于20%,主要原因可能是由于吹扫针在液面上方吹扫不完全。综合考虑,试验选择采用50 mL容量瓶配制后再转移至吹扫瓶中。
试验进一步考察了3种进样方法:3#移液管移取20.00 mL 混合标准溶液至吹扫瓶中;4#移液管移取5.00 mL 混合标准溶液后,吹扫瓶中再加入5.00 mL水;5#移液管移取10.00 mL 混合标准溶液至吹扫瓶中。其中方法3#和5#相比,方法3#部分目标物测定效果较差,这是因为方法3#加入混合标准溶液体积较大,造成吹扫不完全;而方法4#和5#相比,吹扫体积相同,均为10.00 mL,得到的校准曲线经比较,方法5#RRF的RSD(n=8)显著低于方法4#的,这是因为方法5#中加入的混合标准溶液更为均匀,且一次性加入,避免了二次加水引入污染的可能;方法5#与方法2#相比,90%以上目标物的相关系数不小于0.999 0,仅少量在0.998 0~0.999 0内,线性良好,除容易受环境影响的四氯化碳等少数目标物RRF的RSD(n=8)在5.0%~10%内,其余均小于5.0%,说明在液面以下通氮气吹扫比液面以上效率更高。因此,试验选择方法5#进行校准曲线的绘制。
2.2 吹扫时间的优化
在其他条件相同的情况下,考察了吹扫时间(9,11,13,15 min)对氯乙烯、萘回收率的影响,结果如图2所示。
由图2 可知:当吹扫时间由9 min 延长至11 min时,氯乙烯、萘的回收率呈现增高趋势;当吹扫时间由11 min延长至15 min时,氯乙烯的回收率逐渐降低,而萘的回收率继续增高且超于稳定,在13 min后缓慢降低。这可能是由于吹扫时间过长,目标物在未加热脱附前发生部分解吸,被载气带出,导致回收率降低。综合考虑地下水样品和土壤样品,并兼顾不同目标物回收率,11 min均是一个较好的平衡点[3-6,9-10]。因此,试验选择吹扫时间为11 min。

图2 吹扫时间对氯乙烯、萘回收率的影响Fig.2 Effect of purging time on recovery of vinyl chloride and naphthalene
2.3 脱附温度和脱附时间的优化
试验采用Tekmar 9#新型捕集阱(14-9908-403),在查阅相关参考文献基础上,考察了脱附温度(190[4-5,8],200[3,6,9],220[10],250 ℃)对各目标物回收率的影响。结果显示:当脱附温度小于250℃时,各目标物回收率普遍较低;随着脱附温度的上升,各目标物回收率显著增加,但脱附温度过高会缩短捕集阱的寿命。综合考虑,试验选择脱附温度为250 ℃。
在脱附温度250℃条件下及查阅相关参考文献基础上,试验考察了脱附时间(0.5[5,10],1.0,1.5,2.0[14],4.0 min[2,6])对各目标物回收率的影响。结果显示:脱附时间为1.5,2.0 min时,37种VOCs回收率整体较好;但当脱附时间为1.5 min时,萘、六氯丁二烯等高沸点VOCs的回收率小于70%;当脱附时间为2.0 min时,氯乙烯等低沸点VOCs的回收率较差。
因此,进一步选择脱附时间为1.6,1.7,1.8 min进行试验。结果发现,当脱附时间为1.7 min时,各目标物的回收率均达到最高,且高、低沸点VOCs(萘、氯乙烯)的RRF的RSD 均最小。因此,试验选择脱附时间为1.7 min。
2.4 烘烤温度和烘烤时间的优化
Tekmar 9#新型捕集阱的最高使用温度为350 ℃,为了清除残留目标物,烘烤温度应高于脱附温度250 ℃,因此不再考虑215[3],220[4],240 ℃[5]等温度。综合清除效果及吸附剂寿命,试验选择烘烤温度为280 ℃。
在烘烤温度280 ℃条件下,测定目标物质量浓度为40μg·L-1的水样,考察烘烤时间为6,8[8],10 min[3-5,10]对目标物残留的影响,同时测定空白样品。结果表明:烘烤时间为8,10 min时,均可消除样品之间的相互影响。根据节省时间及延长捕集阱使用寿命的原则,试验选择烘烤时间为8 min。
2.5 色谱行为
按照仪器工作条件测定,37种VOCs混合标准溶液的总离子流色谱图见图3。

图3 37种VOCs的总离子流色谱图Fig.3 Total ion chromatogram of 37 VOCs
由图3可知:20 min内可将37种VOCs完全分离,且目标物峰形较好。
2.6 校准曲线与检出限
试验分别采用校准曲线法和RRF 数据处理方法进行定量分析。地下水的线性范围均为1~80μg·L-1,土壤样品中目标物的线性范围均为2~160μg·kg-1,线性回归方程和相关系数的结果见表2。地下水、土壤样品中目标物的RRF 及其RSD(n=8)见表2。
采用HJ 168-2010«环境监测 分析方法标准制修订技术导则»中规定方法计算地下水、土壤样品中目标物的检出限(MDL=3.143sn,s为平行测定的标准偏差,n=7),结果见表2。峰号1~37对应的化合物名称同表1。

表2 地下水和土壤样品中37种VOCs的两种定量方法参数及检出限Tab.2 Parameters of two quantitative methods and detection limits of 37 VOCs in groundwater and soil samples
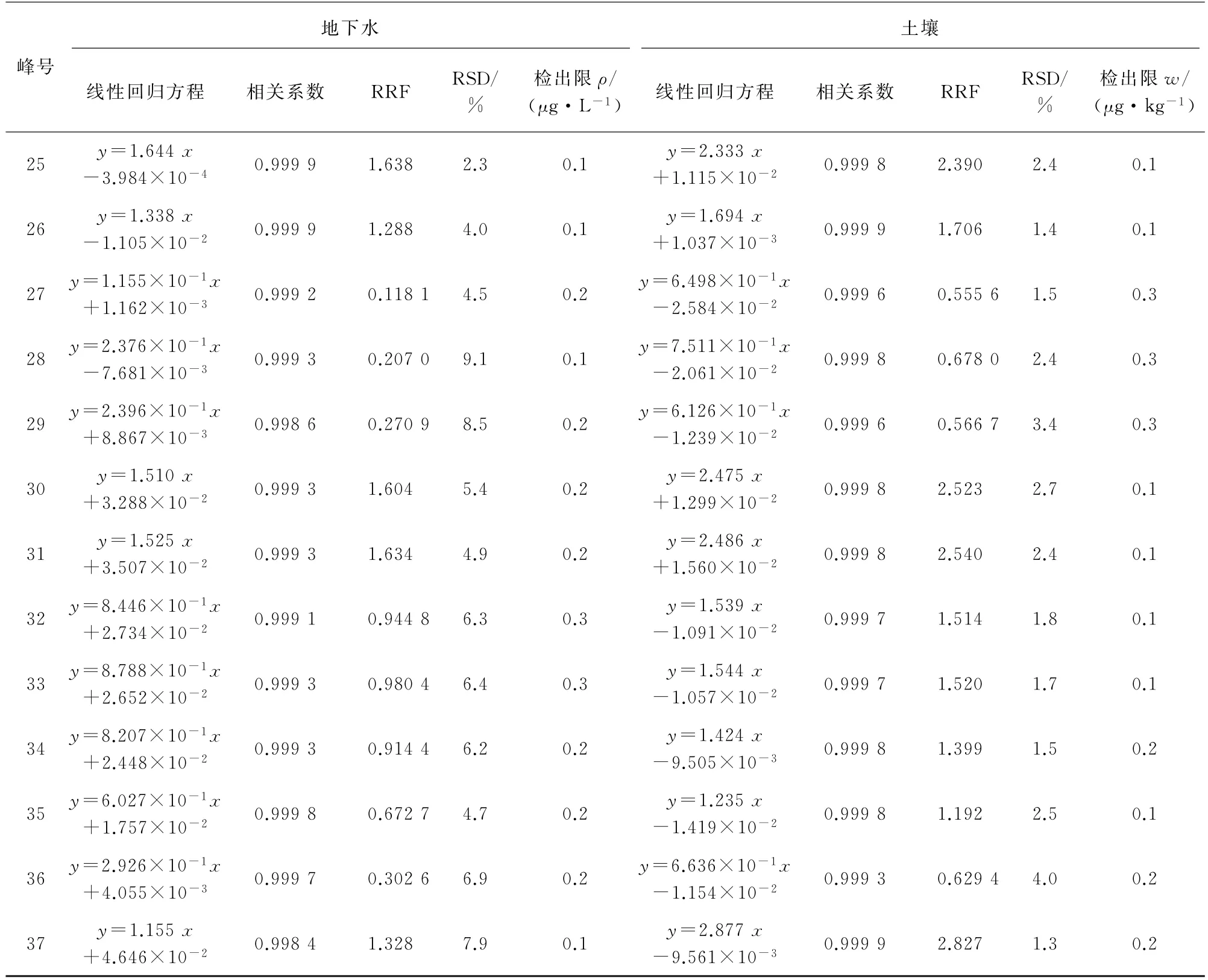
表2 (续)
由表2可知:地下水中各VOCs的检出限均不大于0.5μg·L-1,满足GB/T 14848-2017«地下水质量标准»对Ⅰ类水体的测定要求;土壤中各VOCs的检出限均小于0.4μg·kg-1,远低于GB 36600-2018«土壤环境质量建设用地土壤污染风险管理标准»中最低值0.05 mg·kg-1。两种定量方法可分别满足RRF的RSD 不大于10%及相关系数不小于0.998 0的要求,检出限满足相关标准的测定要求。在实际测定时可选择校准曲线法或RRF数据处理方法,通用性均强。
2.7 精密度和回收试验
以石英砂样品为空白样品,配制4,40,100μg·kg-1等低、中、高3个浓度水平的土壤基体加标样品,同时对加标浓度水平为2,20,40μg·L-1的地下水样进行测定,每个浓度水平平行测定7次,计算回收率和测定值的RSD,结果见表3。
由表3 可知:地下水样中目标物的回收率为75.5%~112%,测定值的RSD 为0.85%~11%,其中低、中、高浓度水平的回收率依次为75.5%~108%,91.0%~109%,85.8%~112%;土壤基体加标样品中目标物的回收率为82.0%~115%,测定值的RSD 为0.81%~7.9%,其中低、中、高浓度水平的回收率依次为83.5%~105%,82.0%~107%,82.2%~115%。回收率随着加标浓度的增大而升高,其中二氯甲烷、四氯化碳的回收率偏低,原因可能是由于两者沸点较低,在测定过程中易损失。

表3 精密度和回收试验结果(n=7)Tab.3 Results of tests for precision and recovery(n=7)
2.8 样品分析
按照试验方法,对济南市7个水源地(S1~S7)的土壤及地下水样品进行分析。其中,仅在部分地下水样品中检出三氯甲烷、四氯化碳和四氯乙烯,结果见表4,而土壤样品中的VOCs均未检出。

表4 样品分析结果Tab.4 Analytical results of samples μg·L-1
本工作采用吹扫捕集-GC-MS快速测定同序列地下水和土壤中37种VOCs的含量,该方法稳定性好,操作简便,在处理数据上具有普适性,适用于地下水和土壤单一介质或同时对VOCs进行批量检测,可为地下水和土壤联合修复提供技术支撑。
猜你喜欢
日用电器(2022年3期)2022-04-14
中国土壤与肥料(2021年5期)2021-12-02
今日农业(2020年22期)2020-12-14
食品安全导刊(2020年33期)2020-12-04
食品安全导刊(2020年21期)2020-12-03
科技创新导报(2020年5期)2020-06-11
食品安全导刊(2017年12期)2018-01-04
科教导刊(2017年26期)2017-11-07
食品界(2017年7期)2017-08-24
科技与创新(2015年17期)2015-09-11
