
脂质特异基质分散萃取超高效液相色谱–串联质谱法测定肉制品中4种兽药残留
2021-08-23 12:37胡雪郢薛丰
化学分析计量 2021年7期
胡雪郢,薛丰
(重庆市渝中区疾病预防控制中心,重庆 400010)
β-受体激动剂(俗称为瘦肉精),曾被用作牛、羊等畜禽的饲料添加剂,其能够促进动物体蛋白质沉积、加快脂肪分解,显著提高脂肪性动物的高饲料转化率(廋肉率)[1],但是科学知识缺乏和经济利益驱使,导致不规范、不合理使用瘦肉精的现象普遍存在。药物滥用会导致药物在动物体内滞留和蓄积,并以残留的方式进入人体以及生态坏境中。误食含瘦肉精的肉类后,会出现头晕、恶心、手脚颤抖、心跳加速甚至心脏骤停,甚致昏迷死亡,对心律失常、高血压、青光眼、糖尿病和甲状腺机能亢进等患者有极大危害[2]。随着全球经济一体化和食品贸易国际化,食品安全已经成为一个世界性的公共卫生问题,而动物源食品违禁药物残留成为全世界关注的焦点[3–4]。为确保食品安全,我国农业部发布的193号公告中明确规定了动物性食品中不得检出β-受体激动剂类药物。
目前,β-受体激动剂残留的检测方法主要有发光法[5–7],气相色谱法[8–10]和质谱法[11–22]。发光法属于快检方法,其操作简单,成本低,但特异性差,灵敏度较低,目前只作为应急快检手段和初筛方法;气相色谱法测定需要进行样品衍生化处理,操作烦琐、结果重现性、可靠性较差,且灵敏度较低,仅能依靠保留时间来进行定性和定量分析等;质谱法通过测定质核比对物质进行定量分析,其特异性和灵敏度较好,气相色谱–质谱法[11–12]应用于瘦肉精残留的测定,与单纯气相色谱法相比,其特异性有所提高,但仍需要对样品进行衍生化反应。液相色谱–质谱法(LC–MS/MS)[13–21]因其灵敏度高、分离性好且操作简便、无需衍生化反应等优势,成为兽药残留检测的首选方法。
肉类食品中兽药残留的检测主要面临两方面的挑战:(1)兽药原型进入动物体内会发生代谢反应,生成一系列的代谢产物,导致原药检测结果的假阴性;(2)动物组织以及内脏中含有大量的蛋白质和脂质,它们性质、含量各异,尤其是大量饱和与非饱和脂肪酸的存在,使得样品基质效应增强;即便通过内标标准曲线[13,21]或者基质曲线来减轻基质效应,也会对测定结果的准确性和重现性产生较大影响,甚至导致方法检出限无法达到限量值要求。
因此,建立高效、快捷的肉类样品中复杂脂质去除前处理方法,是肉类样品检测函待解决的问题。目前,肉类食品中瘦肉精残留检测的前处理方法主要包括:固相萃取(SPE)法[9–10,14–18]和传统基质分散固相萃取(QuEChERS)[19–21]法,后者操作简单、快捷,通用性强,已应用于蔬菜、水果、水产品等样品中农残和兽残的测定,但是对于肉类基质而言,其脂类净化能力有限,不仅无法有效降低基质效应,且在大量样本分析过程中,脂质还会导致色谱和质谱系统发生污染和堵塞,因此,使用新型脂质特异性吸附材料(如免疫亲和萃取法),成为解决此类问题有效途径之一。已有文献报道,将脂质特异性吸附材料用于水产品、烘焙食品中抗生素[22]、抗氧化剂[23]、真菌毒素[24]以及牛肉中多种氨基甲酸酯类农药残留的检测[25],尚未发现在高脂肪含量的猪肉和猪肝样品中β-受体激动剂残留的检测的相关报道。笔者采用脂质特异性吸附EMR–Lipid QuEChER法,结合超高效液相色谱–串联质谱法(UPLC–MS/MS),建立肉类食品中4类β-受体激动剂残留的检查方法,并与已有混合阳离子交换(MCX)固相萃取法进行对比,结果证明,脂质特异性EMR–Lipid QuEChER方法能够更为有效的去除肉类样品中的脂质成分,降低基质效应,重复性好,能够满足日常检测工作需求。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱–串联质谱仪:Dionex UltiMate 3000/AB Sciex 3200 Qtrap型,美国应用生物系统公司。
全自动固相萃取仪:GX–274型,美国吉尔森公司。
恒温震荡培养箱:ZWY–100H型,上海智城分析仪器制造有限公司。
高速冷冻离心机:AvantiJ–26XP型,美国贝克曼库尔特公司。
电子天平:BL310型,感量值为0.01 g,德国赛多利斯公司。
电子天平:XSR204/AC型,感量值为0.1 mg,梅特勒–托利多国际有限公司。
氮吹仪:N–EVAP型,美国Organomation公司。
涡旋混匀仪:VORTEX 3型,艾卡(广州)仪器设备有限公司。
增强型脂质去除分散SPE管:Bond Elut EMR–Lipid型,美国安捷伦科技公司。
β-葡萄糖醛酸苷肽酶/芳基磺酸脂酶:纯度为99%,德国默克公司。
尼龙滤膜:0.22 μm,德国默克公司。
甲醇、甲酸:质谱纯,美国赛默飞世尔科技有限公司。
乙酸–乙酸钠缓冲溶液:优级纯,0.2 mol/L,pH 5.2,美国希格玛公司。
氨水:优级纯,美国赛默飞世尔科技有限公司。
实验用水为超纯水,电阻率大于18 MΩ·cm,由Mili–Q超纯水系统纯化制得。
4种β-受体激动剂标准品以及对应同位素内标标准品基本信息见表1。

表1 4种β-受体激动剂标准品以及对应同位素内标标准品
1.2 溶液配制
一级混合标准溶液:分别取4种β-受体激动剂标准品各100 μL于10 mL容量瓶中,用50%甲醇溶液稀释定容,得到一级混合标准溶液,4种β-受体激动剂的质量浓度均为1.0 μg/mL。
二级混合标准溶液:准确吸取一级混合标准溶液100 μL,用50%甲醇稀释至1 mL,得到二级混合标准溶液,4种β-受体激动剂的质量浓度均为100 ng/mL。
内标储备液:分别用1 mL 50%甲醇溶解1.00 mg D6-莱克多巴胺、1.00 mg D3-沙丁胺醇和1.00 mg D9-特布他林,得到质量浓度均为1.00 mg/mL的D6-莱克多巴胺、D3-沙丁胺醇、D9-特布他林的内标储备液。用2.13 mL 50%甲醇溶解2.40 mg D9-盐酸克伦特罗,配制成质量浓度为1.00 mg/mL的D9-盐酸克伦特罗内标储备液。其中,D9-盐酸克伦特罗的质量浓度以D9-克伦特罗计,换算系数为88.8%。
混合内标应用液:分别取4种同位素内标储备液各10 μL于10 mL容量瓶中,用50%甲醇溶液稀释定容,得到4种同位素内标混合应用液,质量浓度为1.0 μg/mL。
内标系列标准工作溶液:分别准确吸取二级混合标准溶液5、10、20、50 μL,以及一级混合标准溶液10、30、50 μL,分别加入30 μL内标混合应用液,再用超纯水稀释至1 mL,混匀。4种化合物标准系列溶液浓度依次为:0.5、1.0、2.0、5.0、10、30、50 ng/mL;4种内标化合物的浓度均为30.0 ng/mL。
1.3 仪器工作条件
1.3.1 色谱条件
色谱柱:Welch Xtimate C18型柱[100 mm×2.1 mm,3 μm,月旭科技(上海)股份有限公司];柱温:40 ℃;流量:300 μL/min;进样体积:10 μL;流动相:A相为0.1%甲酸,B相为甲醇,梯度洗脱程序列于表2。
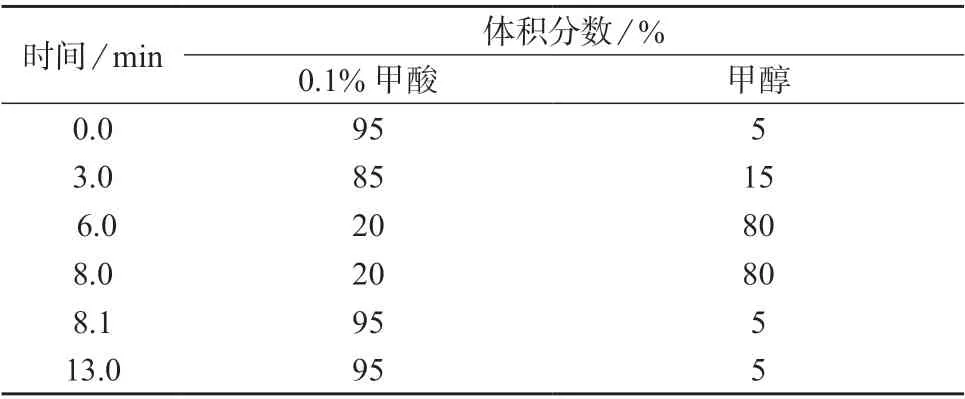
表2 梯度洗脱程序
1.3.2 质谱条件
电离方式:ESI+;喷雾电压:5 000 V;离子源温度:500 ℃;雾化气(Gas l)流量:50 mL/min;辅助加热气(Gas 2)流量:50 mL/min;气帘气流量:25 mL/min;碰撞气流量:6 mL/min;监测模式:多反应监测模式(MRM)。
1.4 样品前处理
1.4.1 样品提取
称取5 g(精确到0.01 g)粉粹的样品于50 mL离心管中,加入30 μL 1.0μg/mL的混合内标应用液,再加入15 mL 0.2 mol/L pH 5.2乙酸–乙酸钠缓冲溶液,均质器匀浆后,再加入100 μLβ-葡萄糖醛酸苷肽酶/芳基磺酸脂酶,于(37±1)℃震荡酶解过夜,然后于4 ℃以12 000 r/min离心10 min,取上清液待净化。
1.4.2 样品净化
(1)混合型强阳离子交换反相吸附(MCX)固相萃取法。取Oasis MCX型固相萃取小柱[(150 mg)/3 mL,美国沃特世公司],经40 mmol/L HCl水溶液活化后,取上清液全部上样,分别用5 mL水和甲醇淋洗,再用6 mL 5%氨水–甲醇分3次洗脱,每次2 mL。合并洗脱液,经氮气吹干后,用甲醇–0.1%甲酸(5∶95)定容至1.0 mL,用0.22 μm滤膜过滤,采用UPLC–MS/MS法测定。
(2)增强型脂质去除(EMR–Lipid QuEChERS)法。移取上清液至EMR–Lipid d-SPE管中,然后加入等体积的甲醇,立即涡旋混合使样品分散,涡旋60 s后以5 000 r/min离心3 min,移取5 mL上清液至含有2 g 盐(氯化钠与硫酸镁质量比为1∶4)的15 mL EMR–Lipid Polish管中,漩涡混合1 min后,以5 000 r/min离心3 min ,上清液经氮气吹干后,用甲醇–0.1%甲酸(5∶95)定容至1.0 mL,用0.22 μm滤膜过滤,采用UPLC–MS/MS法测定。4种β-受体激动剂标准色谱图见图1,其同位素内标标准色谱图见图2。
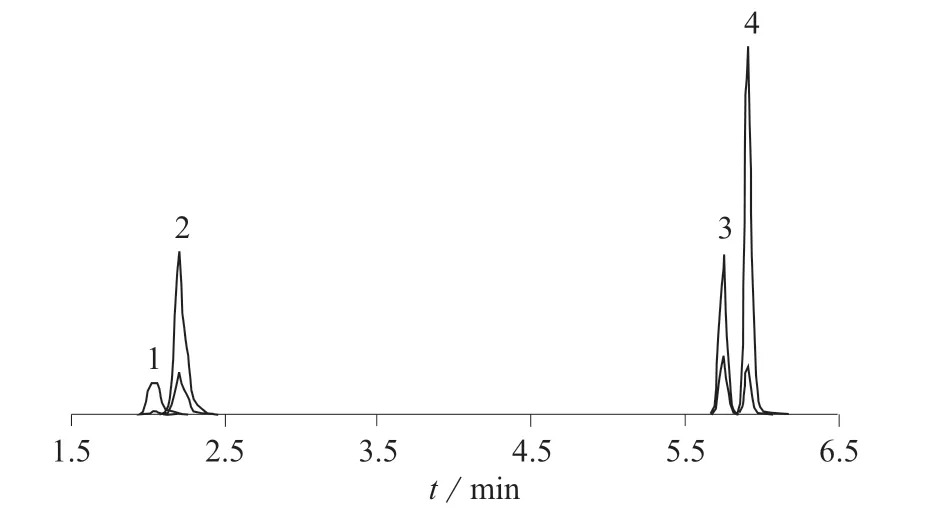
图1 4种β-受体激动剂标准色谱图
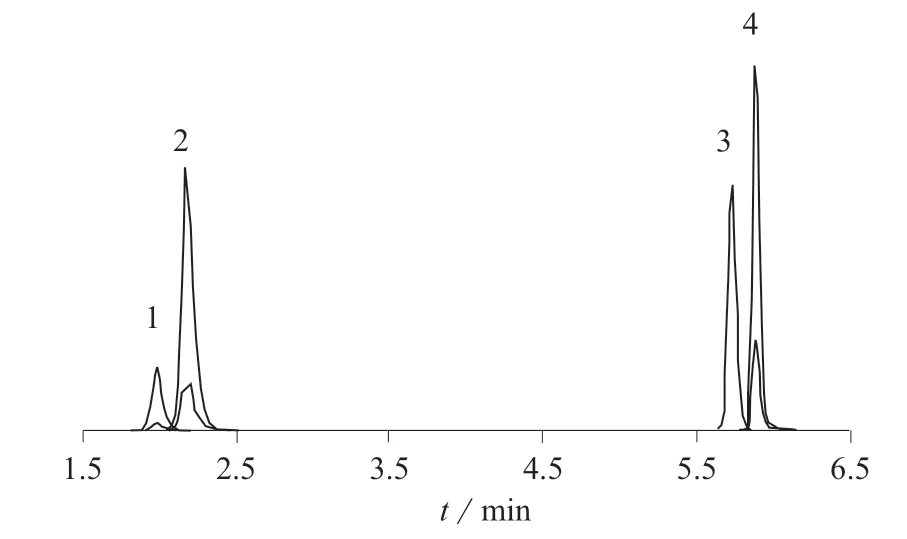
图2 4种β-受体激动剂同位素内标标准色谱图
2 结果与讨论
2.1 4种β-受体激动剂动物体内的存在形式研究
药物进入动物体内,会经过一系列的酶促反应之后排出体外,反应中将生成一系列的代谢产物,由于药物理化性质的差异以及作用位点不同,其代谢场所各异,其中参与药物代谢比较活跃的组织器官包括肠道、肝脏、肾脏以及肌肉组织。本研究通过体外培养模拟肝脏和肌肉组织中4种β-受体激动剂的代谢反应,研究4种化合物在上述动物组织中的存在形式。
称取5.0 g新鲜猪肉和猪肝各两份,设置实验组和对照组,每组含猪肉和猪肝样品各1份;均加入1 mL 50 ng/mL的内标标准曲线系列溶液,37 ℃恒温培养24 h,实验组培养过程中不加入β-葡萄糖醛酸苷肽酶/芳基磺酸脂酶,对照组则完全按照1.4.1标准流程进行操作,培养完成后所有样品按照1.4.2(1)方法进行样品净化后测定。结果表明,4种化合物在猪的肌肉和肝脏中几乎都以化合物原型存在。
2.2 离子对选择和质谱参数优化
将一级混合标准溶液和混合内标应用液等体积混合,通过针泵直接进样方式,对每种化合物的母离子和子离子进行选择,同时分别优化其质谱采集参数:碰撞电压(CE)、去簇电压(DP)、入口电压(EP)和碰撞池出口电压(CXP),优化结果列于表3。
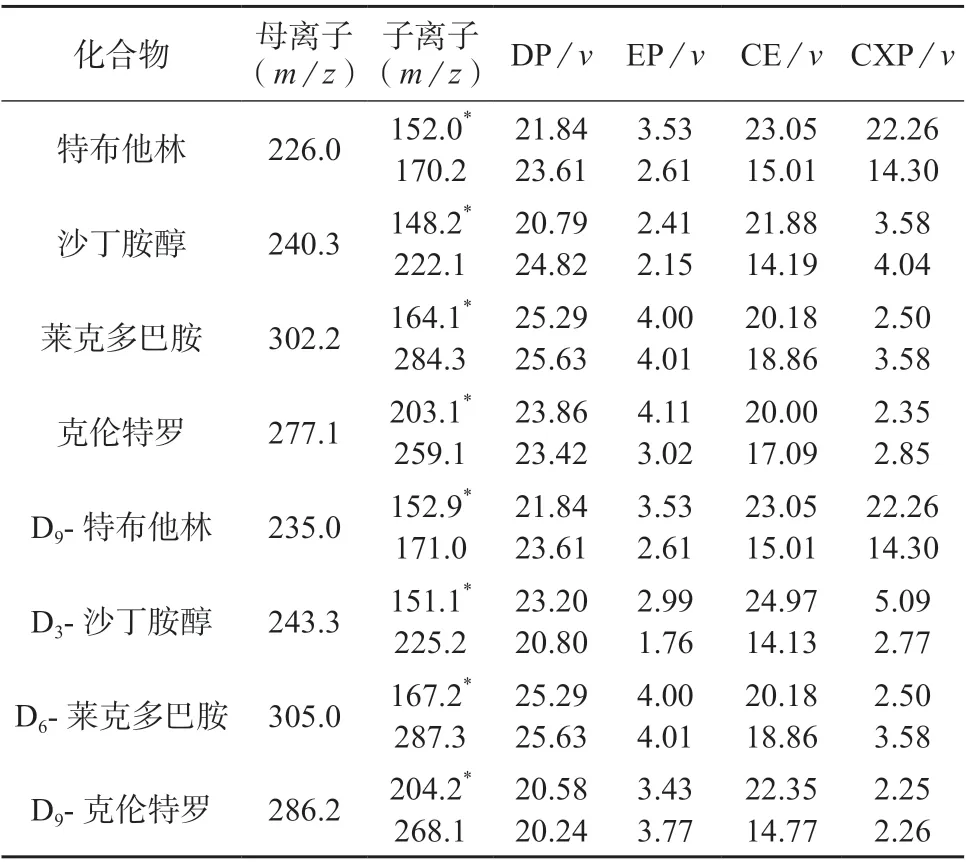
表3 4种β-受体激动剂及其对应内标物质监测离子、质谱参数
2.3 前处理方法比较
动物组织样品进行分析时,大量脂质的存在是干扰分析结果准确性的主要原因。动物所含脂质的分子量较大(含碳原子数≥6),且多为直链结构,支链较少,EMR–Lipid QuEChERS中的脂质特异性吸附填料通过其自身特殊的多孔构象,能够对长直链脂肪进行基于空间位阻理论的特异性吸附。本研究以1.4.2中混合型强阳离子交换反相吸附(MCX)固相萃取法为参照,与EMR–Lipid QuEChERS法进行比较,通过相同基质(肌肉组织和肝脏)提取液进行净化处理,对样液进行一级质谱(MS1)100~850m/z质量范围内全扫描分析。结果表明,选择C18填料的色谱柱、0.1%甲酸水溶液和甲醇作为流动相的色谱分离体系中,脂质的保留能力较强,其色谱流出时间位于流动相中有机相比例较高的时间段(5~7 min),在此时间段内,经MCX处理的样液中有明显杂质色谱峰出现。与MCX法相比,EMR–Lipid QuEChERS法对猪肉以及猪肝中脂质去除效果更好。
2.4 线性方程与检出限
为了尽量减少基质效应的影响,采用内标标准曲线法进行定量,以4种β-受体激动剂类药物的色谱峰面积(x)为横坐标,质量浓度(y)为纵坐标绘制标准曲线,依据信噪比S/N=3计算方法的检出限。各化合物线性回归方程、相关系数、检出限列于表4。结果表明,4种化合物的质量浓度在0.5~50 ng/mL范围内线性关系良好,相关系数均大于0.997,检出限均为0.1 μg/kg。

表4 4种β-受体激动剂类药物的线性方程、相关系数、检出限
2.5 精密度与加标回收试验
取经过检测的空白新鲜猪肉和猪肝样品,选取2.0、5.0、10.0 ng/kg 3个加标水平,每个浓度水平平行测定6次,按照1.4.2(2)中优化的实验方法进行分析,计算目标化合物的回收率和相对标准偏差(RSD),结果列于表5和表6。由表5和表6可知,4种β-受体激动剂在猪肉基质中的加标回收率为79.8%~92.3%,测定结果的相对标准偏差为2.95%~9.50%;在猪肝中的加标回收率为81.5%~95.6%,测定结果的相对标准偏差为2.47%~8.05%。表明该方法具有较高的准确度和精密度,满足日常检测需求。

表5 4种β-受体激动剂类在猪肉样品中加标回收试验结果
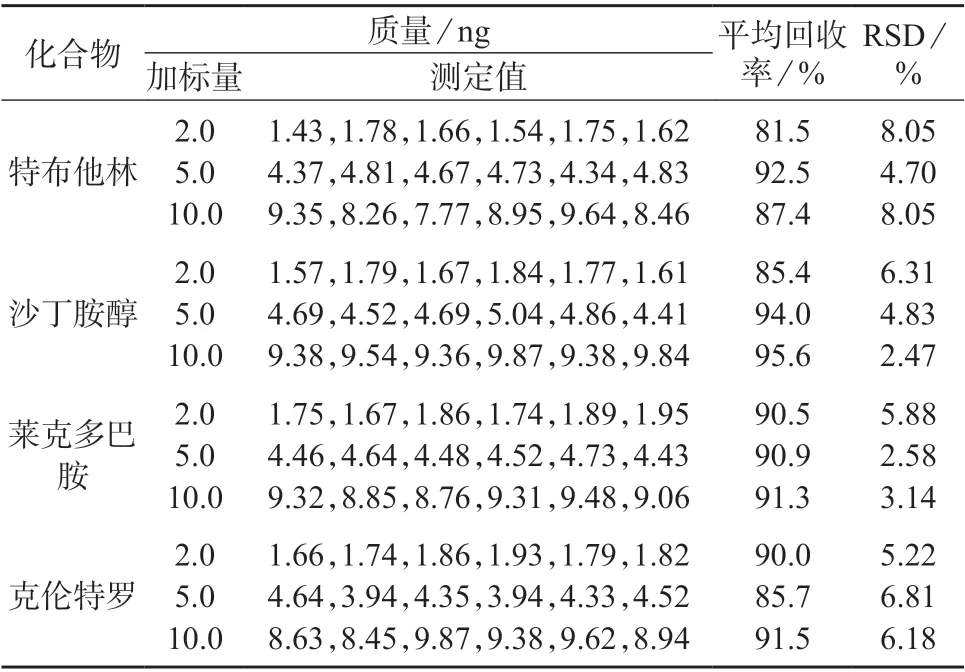
表6 4种β-受体激动剂类在猪肝样品中加标回收试验结果
2.6 实际样品测定
将本研究所建立的脂质特异性基质分散(EMR–Lipid QuEChERS)净化结合超高效液相色谱–串联质谱法对市售的34个肉类样品(包括猪肉、猪肝、鸡肉、牛肉、鱼肉5个大类)进行测定,发现未检出克伦特罗、莱克多巴胺、沙丁胺醇、特布他林的违法添加。
3 结语
脂质特异性基质分散(EMR–Lipid QuEChERS)净化法用于肉类样本中脂质的去处,采用类似QuEChERS标准样品前处理流程,相比传统固相萃取法,操作更加简便、灵活和快捷,且脂质去处效果也更好,结合超高效液相色谱–串联质谱法的高特异性和灵敏度,能够满足日常工作中兽药残留的检测工作。由于EMR–Lipid QuEChERS能够特异性得吸附脂质(尤其是含6个C以上的长链脂肪),而非对待测物进行吸附和富集,因此,此法也可应用于其他高油脂样品中脂质的去除,从而降低机制效应的干扰,提高目标化合的检出限以及定量准确性。
猜你喜欢
浙江化工(2022年1期)2022-02-19
现代临床医学(2021年6期)2021-11-20
口腔护理用品工业(2021年4期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
天津医科大学学报(2021年3期)2021-07-21
昆明医科大学学报(2021年1期)2021-02-07
中华养生保健(2020年9期)2021-01-18
中成药(2019年12期)2020-01-04
自我保健(2019年10期)2019-12-11
中国实验动物学报(2019年4期)2019-09-03
