
儿童脑型X-连锁肾上腺脑白质营养不良的临床特点及基因突变特征
2021-08-20 10:01赵盼丁金金王越华爽刘念麦子荆田培超
河南医学研究 2021年19期
赵盼,丁金金,王越,华爽,刘念,麦子荆,田培超
(郑州大学第一附属医院 儿科,河南 郑州 450052)
X-连锁肾上腺脑白质营养不良是一种过氧化物酶体病,是X连锁先天遗传性疾病,临床较罕见,X-连锁肾上腺脑白质营养不良包含一系列表型,包括脑型(儿童、青少年和成人)、肾上腺脊髓神经病型、单纯Addison病型、无症状型和杂合子型[1-2]。脑型中以儿童脑型最为常见,且病情进展快,存活时间短,病死率高。本研究回顾性分析郑州大学第一附属医院收治的11例脑型X-连锁肾上腺脑白质营养不良患儿的临床特点及基因突变特征,为早期诊断儿童脑型X-连锁肾上腺脑白质营养不良提供参考。
1 资料与方法
1.1 一般资料对2015年1月至2020年6月郑州大学第一附属医院确诊的11例脑型X-连锁肾上腺脑白质营养不良患儿的临床特点及基因突变特征进行分析。依据其典型的临床表现、家族史、实验室检查、头颅影像学及基因突变结果确诊。未纳入青少年和成人脑型、肾上腺脊髓神经病型、单纯Addison病型、无症状型和杂合子型。
1.2 研究方法收集11例患儿临床表现、实验室检测及头颅影像学检查结果。采集10例患儿静脉全血标本进行高通量测序,采用Sanger测序法对患儿父母及兄弟姐妹ABCD1基因突变进行验证。检索HGMD数据库判断患儿基因突变结果是否为新突变,对于突变基因的致病性按照基因组学学会指南或通过PolyPhen-2软件预测是否致病。
2 结果
2.1 临床特征基本资料:11例患儿均为男性,起病年龄2~9岁,中位年龄6岁,表现为视力下降9例,智力低下5例,步态不稳4例,性格改变3例,多动、注意力不集中3例,运动及体格发育落后1例,癫痫发作1例,听力障碍1例,肢体无力1例,皮肤和黏膜色素沉着增加2例。阳性体征:9例直接或间接对光反射迟钝,伴有不同程度视力下降及眼球运动障碍,4例膝腱反射亢进、Babinski征阳性,3例双下肢肌张力增高、痉挛步态,2例体格发育迟缓、全身皮肤黏膜色素沉着,2例Chaddock征阳性,2例腹壁反射消失。
2.2 实验室及影像学资料本研究中3例患儿肾上腺皮质功能不全,实验室检测结果显示血清促肾上腺皮质激素升高、皮质醇降低或促肾上腺皮质激素兴奋试验无反应。对6例患儿进行血清极长链脂肪酸(very long chain fatty acid,VLCFA)测定,血清二十六烷酸(hexacosanoic acid,C26∶O)、二十四烷酸(tetracosanoic acid,C24∶O)/二十二烷酸(behenic acid,C22∶O)、C26∶O/C22∶O均有不同程度的升高。对11例患儿行头颅MRI平扫检查,均可见双侧对称性以顶枕叶为主的蝴蝶翼状脑白质异常信号,其中9例累及双侧顶叶,9例累及双侧枕叶、侧脑室后角旁,8例累及胼胝体压部,7例累及双侧大脑脚,5例累及丘脑、脑桥,4例累及双侧颞叶、额叶、基底节区,3例累及双侧侧脑室前角,2例累及脑干、小脑中脚、双侧侧脑室三角区,1例累及放射冠,1例累及双侧侧脑室颞角旁。
2.3 基因检测结果10例患儿行基因测序发现均有ABCD1基因突变,其中9例为错义突变,1例为移码突变。见表1。查阅相关文献报道及基因库,其中编号为1和3患儿的基因突变是目前HGMD数据库尚未收录的新突变。患儿1的突变位点为c.1161_1174del(图1),患儿3的突变位点为c.1931T>C。上述变异经ACMG致病性分级评定为致病,PolyPhen-2预测为有害。
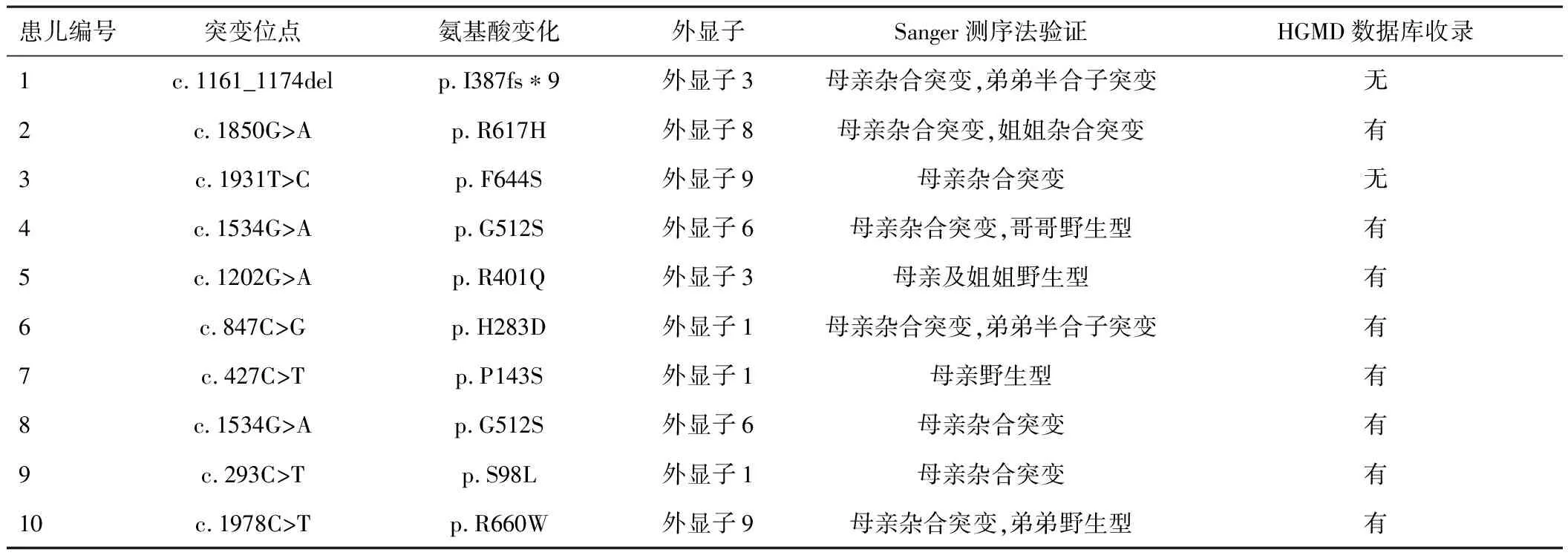
表1 10例患儿基因测序结果
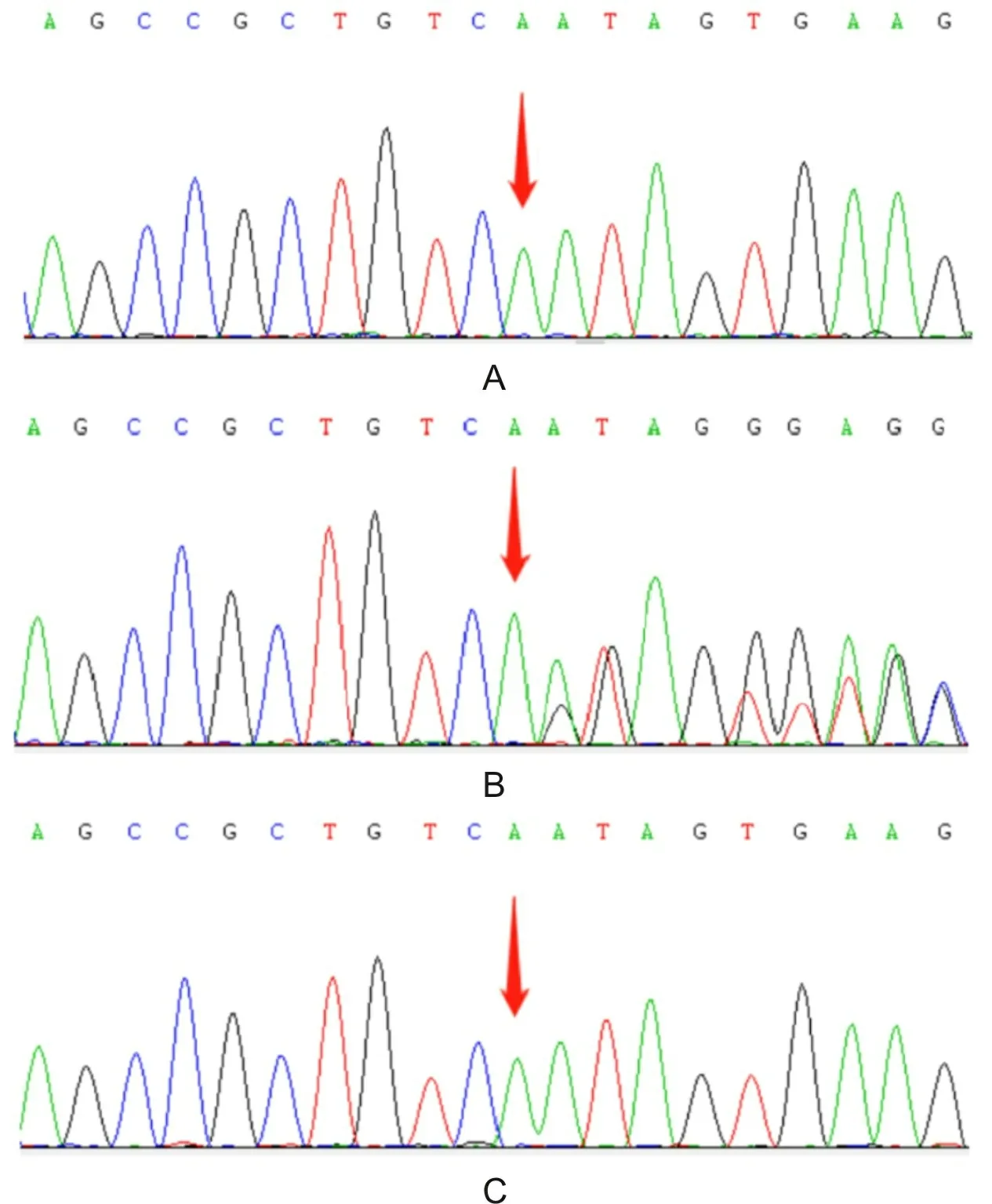
A为先证者在chrX:153001645位置半合子突变;B为先证者母亲在chrX:153001645位置杂合突变;C为先证者弟弟在chrX:153001645位置半合子突变。
2.4 治疗及预后给予3例肾上腺皮质功能不全患儿氢化可的松口服治疗后肾上腺皮质功能不全有所缓解,神经系统症状仍持续性加重。给予2例患儿罗伦佐油及饮食指导,神经系统症状仍持续进展。随访6个月~5 a,5例存活患儿在6个月~1 a内病情迅速进展,基本完全失能,出现痉挛性截瘫、失明、失聪、失语、癫痫发作,6例患儿在发病1~4 a后死亡。
3 讨论
X-连锁肾上腺脑白质营养不良是一种进展性疾病,预后多数取决于其表型,脑型X-连锁肾上腺脑白质营养不良约占X-连锁肾上腺脑白质营养不良的35%,在儿童期最常见,进展速度快,且病死率高,通常4~10岁起病,发病高峰年龄7岁。发病初期通常表现为行为变化、多动、注意力下降、进行性智力及运动能力减退、写字困难、视力障碍、失聪、失语。个别患儿可以抽搐为首发症状。儿童脑型X-连锁肾上腺脑白质营养不良症状一旦发生可在数周内迅速恶化,多在2~4 a发展至植物状态或死亡。部分患者同时伴有肾上腺皮质功能低下的临床表现[3]。头颅MRI检查多会发现以顶枕区为主的双侧对称性蝴蝶翼状脑白质异常信号[4],与本研究结果相符。
X-连锁肾上腺脑白质营养不良是由位于Xq28的ABCD1基因发生突变导致的,ABCD1基因包含10个外显子和9个内含子,长约21 kb[5]。肾上腺脑白质营养不良突变数据库目前共收录2 901个ABCD1基因突变位点,其中以错义突变最为常见,约占62%,其次为移码突变,约占16%(http://www.x-ald.nl/)。ABCD1基因编码ATP结合盒(ATP-binding cassette,ABC)转运蛋白,ABC转运蛋白是一种广泛分布的膜蛋白,其功能是帮助VLCFA进入过氧化物酶体通道进行β氧化[6]。ABCD1基因突变可能导致ABC转运蛋白功能障碍,阻碍VLCFA正常转运进入过氧化物酶体,从而阻止VLCFA的β氧化和降解,体内升高的VLCFA在中枢神经系统、肾上腺皮质等受累器官蓄积是X-连锁肾上腺脑白质营养不良病理改变的基础[7-8],而VLCFA在中枢神经系统的蓄积导致星形胶质细胞、少突胶质细胞及小胶质细胞的神经炎症反应[9-11]。本研究11例患儿中有10例行ABCD1基因检测,结果发现均有基因突变,其中9例为错义突变,1例为移码突变,导致其编码蛋白质的氨基酸改变或编码提前终止,致ABC转运蛋白失去原有的功能,1例未行基因检测的患儿有阳性家族史。本研究中2例患儿出现了新突变:患儿1的ABCD1基因第1 162~1 173位碱基缺失导致ABC转运蛋白第387位的异亮氨酸由于移码突变的发生,从第387位开始计数的第9位变为终止密码,经ACMG致病性分级评定为致病,PolyPhen-2预测为有害;患儿3的ABCD1基因由于第1 931位碱基T被碱基C取代,导致第644位氨基酸由苯丙氨酸变成丝氨酸,经ACMG致病性分级评定为致病,PolyPhen-2软件预测为有害。本研究发现的2例新突变丰富了ABCD1基因突变谱。
研究证明,罗伦佐油有降低血清VLCFA水平的效果,但对神经系统的疗效仍需进一步研究[12]。药物治疗目前多在研究中,包括上调ABCD2表达水平的药物及作用于GSK-3β/Nrf2轴的Nrf2激动剂等[13]。对伴有肾上腺皮质功能不全的患儿可应用皮质类固醇激素替代疗法,本研究中有3例患儿出现肾上腺皮质功能不全,均口服氢化可的松治疗。异基因造血干细胞移植是目前可选择的有效治疗手段,其治疗肾上腺脑白质营养不良的机制暂不明确,有研究认为其可能机制是供者提供的骨髓来源的小胶质细胞具有正常的溶酶体功能,从而阻止神经系统病变的进展[14]。目前研究认为处于病程早期的患儿比较适合接受造血干细胞移植,早期的定义为没有或仅有1项神经系统缺陷(不包括认知和行为症状)且头颅MRI异常为轻度,MRI结果的严重程度评分<9分[15]。目前许多X-连锁肾上腺脑白质营养不良患儿缺乏人类白细胞抗原完全相合的亲缘或非亲缘供体,而单倍体配型相同的亲缘供体可以使缺乏合适供体的患儿及时获得供者造血干细胞。文献报道,单倍体相合亲缘供体很容易获得,这可以减少诊断和移植之间的时间间隔,发挥移植治疗的积极效果[16]。单倍体相合异基因造血干细胞移植治疗肾上腺脑白质营养不良是可行的,但移植的长期疗效还需要进一步观察[17]。在疾病早期成功进行造血干细胞移植的男性患儿,预后一般良好,5 a生存率可高于90%,但造血干细胞移植并不能治愈该病,在成年期可能出现脊髓病症状[18]。目前正在研究的基因疗法或许能带来新的希望,主要是应用重组病毒相关载体将正常ABCD1基因转染至中枢神经系统,目前已开展临床试验,且取得了较好的疗效,但需要长期的随访,全面评估其疗效[19]。
北美X-连锁肾上腺脑白质营养不良患儿的出生率为1∶17 000,为避免X-连锁肾上腺脑白质营养不良患儿的出生,产前检查十分重要[20]。对于娩出X-连锁肾上腺脑白质营养不良患儿、家族史阳性及基因检测为杂合子的女性,可进行产前检测。产前诊断可以在女性妊娠时取胎盘绒毛或羊水细胞,通过ABCD1基因分析进行胎儿诊断,避免X-连锁肾上腺脑白质营养不良患儿的出生。进行辅助生殖技术时,可采用多重置换扩增对胚胎完成植入前胚胎遗传学诊断。新生儿筛查目前多采用C26∶O溶血磷脂酰胆碱高通量串联质谱分析[21]。对于筛查结果为阳性的婴儿,应尽快进行血清VLCFA和ABCD1基因突变分析来确诊。基因检测结果显示为杂合子的女性在妊娠时需采集羊水细胞进行ABCD1基因分析,基因检测结果显示为半合子的男性需进一步完善血清VLCFA测定及头颅影像学检查,在疾病早期进行治疗,可延缓病情进展,改善预后。
综上所述,X-连锁肾上腺脑白质营养不良是一种罕见遗传代谢病,临床表型多样,儿童脑型较多见,临床表现主要为进行性听力和视力下降、智力和运动能力减退及肾上腺皮质功能不全,查体可见神经系统阳性体征。检测可发现血清VLCFA水平升高。其头颅影像学检查多见双侧对称性脑白质病变,主要累及双侧顶叶、枕叶、侧脑室后角旁、胼胝体压部、大脑脚、丘脑及脑桥等,基因检测可确诊。本研究发现2个ABCD1基因新突变,丰富了ABCD1基因突变谱。目前尚无特效的治疗方法,造血干细胞移植可延缓病情进展,但其长期疗效还需要进一步观察,基因治疗或可成为新的希望。
猜你喜欢
天津医科大学学报(2021年4期)2021-08-21
昆明医科大学学报(2021年3期)2021-07-22
中国生殖健康(2020年2期)2021-01-18
小资CHIC!ELEGANCE(2021年46期)2021-01-11
睿士(2020年11期)2020-11-16
VOGUE服饰与美容(2020年9期)2020-09-02
医学新知(2019年4期)2020-01-02
保健与生活(2019年15期)2019-09-12
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
