
丙烯酰乙二胺-β-环糊精毛细管电色谱整体柱的制备及应用
2021-08-09 09:36:56唐艺旻李英杰高立娣秦世丽靳凤龙
黑龙江大学自然科学学报 2021年3期
唐艺旻, 李英杰, 高立娣, 秦世丽, 赵 冰, 靳凤龙
(1.齐齐哈尔大学 化学与化学工程学院, 齐齐哈尔 161006; 2.齐齐哈尔市场监督管理局食品药品检验检测中心, 齐齐哈尔 161005)
0 引 言
手性广泛存在于自然界中,手性药物对映体三维空间结构之间的差异可导致不同的药理学和毒理学差异[1-2]。手性药物约占全球药物销售量的1/3,这些手性药物对映体中的一种对映体可能产生预期的药理学反应,而相反的一种可能只表现出拮抗或毒性反应[3]。正是对映体药理活性的重要差异,使得手性药物对映体的拆分逐渐受到重视。
高效液相色谱(High performance liquid chromatography, HPLC)[4-6]、气相色谱(Gas chromatography, GC)[7-8]、毛细管电泳(Capillary electrophoresis, CE)[9]和毛细管电色谱(Capillary electrochromatography, CEC)[10]等经常用于对映体分离。但HPLC与GC所使用的手性柱价格昂贵,且大多数情况下,一个特定的手性柱只能分析某一类物质,应用往往受限。CEC不仅具有CE的高效性,而且兼具HPLC的高选择性,成为一种选择性好、分离效率高、消耗试剂量小的分离分析技术[10]。β-环糊精(β-CD)及其衍生物由于空腔大小适中、良好的柔韧性和天然的手性已被广泛使用在CEC和CE领域中[11]。Zhou等开发了一种溶胶-凝胶“一步法”来制备SBE-β-CD-二氧化硅杂化整体柱,整体柱内固定相具有均一的结构。优化色谱条件后,在CEC中共分离了26种外消旋分析物,其中17种达到了基线对映体分离[12]。Deng等首次通过一锅法制备了羟丙基-β-环糊精(HP-β-CD)功能化的毛细管整体柱,通过NMR和拉曼光谱确认了HP-β-CD在整体柱上成功键合,在CEC上成功分离外消旋化合物,包括外消旋抗胆碱能药物、甲哌嗪醇及其中间体等[13]。
本文旨在合成一种具有多个作用位点、制备简单的β-CD衍生物,6-EA-β-CD在β-CD基础上引入双键和胺基等基团,不仅克服了β-CD水溶性差的缺点,而且增强了氢键和包结等作用。利用CEC的高效性和高选择性,对手性化合物进行拆分,为手性拆分领域提供一些基础数据。
1 实验部分
1.1 仪器与试剂
7100 CE毛细管电泳仪(美国安捷伦公司)、6230 TOF MS-质谱仪(美国安捷伦公司);S-4300 SEM(日本日立公司);Nicolet 380 FTIR傅立叶变换红外光谱仪(美国热电公司);DMBA 200 光学显微镜(麦克奥迪医疗诊断有限公司);石英毛细管柱(75 μm i.d., 365 μm o.d., 中国锐丰色谱器材公司)。
β-环糊精、N,N-二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)、1,3-二环己基碳化二亚胺(DCC)(天津科密欧化学试剂有限公司);偶氮二异丁腈(AIBN)、2-丙烯酰胺基-2-甲基丙磺酸(AMPS)、二甲基丙烯酸乙二醇酯(EDMA)(分析纯,阿拉丁试剂);盐酸班布特罗(中国天津中央药业有限公司)。
1.2 实验方法
1.2.1 单6-丙烯酰乙二胺-β-环糊精(6-EA-β-CD)的合成
首先合成磺酰化-β-环糊精(6-OTs-β-CD)[14]和单6-乙二胺-β-环糊精(6-en-β-CD)[15],然后称取2 g 6-en-β-CD和0.36 g DCC,溶解于20 mL DMF中,控制反应温度15 ℃下滴加0.34 g丙烯酸,30 min后,室温下反应8 h。反应结束后真空抽滤,滤液用一定量的丙酮析出沉淀,真空干燥,得浅黄色固体产物 6-EA-β-CD。
1.2.2 整体柱的制备

1.3 电色谱柱性能研究
以D,L-丙氨酸和盐酸班布特罗为分析物,对6-EA-β-CD整体柱的手性拆分性能进行了研究。在毛细管电色谱运行过程中,以20 mmol·L-1醋酸铵作为缓冲液,50 mbar×3 s压力进样,紫外检测波长为200 nm。
同时采用单一变量法以分离度为考察指标,优化考察影响分离的主要因素,包括缓冲溶液的pH值、电压和柱温,如表1所示,从而得出最佳电色谱分离条件。
2 结果与讨论
2.1 6-EA-β-CD的表征
2.1.1 红外光谱分析

表1 CEC分离条件的影响因素

图1 红外光谱图: (a)磺酰化-β-环糊精; (b)乙二

图2 6-EA-β-CD 的质谱图
2.1.2 质谱分析
采用ESI-TOF/MS对合成产物进一步验证,结果如图2所示。6-EA-β-CD分子量为1 230,图2中m/z为1 231.453 63的离子峰对应6-EA-β-CD的[M+H]+峰,m/z为1 253.483 71的离子峰对应[M+Na]+峰,m/z为1 269.398 5是6-EA-β-CD的 [M+K]+峰,从而可以断定合成的环糊精衍生物为6-EA-β-CD。
2.2 整体柱制备条件的优化
采用一步键合法制备的整体柱制备简单、环保,具有较好的重现性以及较大的固定相承载能力[16]。当6-EA-β-CD单体溶液的浓度为0.15 g·mL-1、EDMA功能单体的量为0.060 g时,保证总体系中致孔剂质量百分数为 86%。分别采用光学显微镜和甲醇经高压恒流泵流入整体柱中的流速为指标评价整体柱的键合情况和通透性,结果如表2所示。

表2 反应温度及时间对柱性能的影响
从表2光学显微镜观察到的结果可以看出,当反应时间不变时,随着反应温度的升高,柱内固定相的键合情况从稀疏变得紧密。结合甲醇流速可以看出,当温度为45 ℃时,甲醇流速适中,柱内通透性较好,从而确定最佳反应温度为45 ℃。反应时间也是整体柱内固定相键合情况的重要影响因素,一方面反应时间过少,固定相键合不紧密,溶质的保留较小;另一方面,若反应时间太长,柱内的团聚现象明显,柱子通透性下降。当反应温度均为45 ℃、反应时间为6和8 h时,柱内固定相键合量较多且相对均匀,结合甲醇流速可以看出,反应时间为8 h时,甲醇流速较慢,柱子通透性差,而反应时间为6 h时甲醇流速适中,通透性良好,最终确定反应时间6 h为最佳条件。与吕仁江等制备的丙烯酰胺-β-CD毛细管电色谱整体柱[17]相比,两种整体柱均采用一步键合法制备,本文通过改变反应温度和时间,在更短的时间内制得了具有良好性能的整体柱。
2.3 整体柱的扫描电镜表征
整体柱内部固定相形貌采用SEM进行表征,结果如图3所示。图3(a)和图3(b)分别为在最佳制柱条件(6-EA-β-CD单体溶液的浓度为0.15 g·mL-1、EDMA为0.060 g、总体系中致孔剂质量百分数为 86%、反应温度为45 ℃、反应6 h)下制得的整体柱横截面和局部截面扫描电镜图。可以看出,柱内形成了孔径较均匀的网状结构,其中,大中孔径保证了一定的通透性。固定相与柱内壁键合紧密,柱内固定相不会轻易被流动相冲出柱外,整体柱稳定性得以提高。当其他制柱条件不变,控制反应时间为4 h时制得的整体柱横截面如图3(c)所示,柱内固定相键合量较少,柱内稀疏,通透性好;反应时间为8 h时制得的整体柱横截面如图3(d)所示,柱内键合紧密,有明显的团聚现象,柱内孔径较小,通透性差。由此验证,在最佳制柱条件下制得的整体柱具有较好的形貌,柱性能良好,可用于进一步的应用研究。


2.4 整体柱的性能研究
柱效是色谱柱分离能力的度量,通过计算有效塔板数对该整体柱的柱效进行评价。整体柱长度一定时,有效塔板数越大,表明物质在整体柱中分配平衡次数越多,对分离越有利。本文在最佳制柱条件下制备三根电色谱整体柱,以硫脲为中性标记物,对所制备整体柱的柱效进行评价,结果如表3所示。三根柱子平均柱效达到64 251 plates·m-1以上,并且单根柱子柱效的相对标准偏差(RSD)分别为0.728%、0.151%和0.895%。表明该整体柱柱效较高,有一定的分离能力,且柱子相对稳定。

表3 整体柱柱效评价
以D,L-丙氨酸和盐酸班布特罗为分析物,对6-EA-β-CD整体柱的手性拆分性能进行进一步考察。手性化合物拆分过程会受缓冲溶液pH、电压及温度的影响,故采用控制单一变量法对这些条件进行了考察,结果如图4所示。图4(a)~图4(c)分别为缓冲溶液pH值、电压和温度对分析物分离度的影响,从图4中可以看出,当pH值为8.5、电压为20 kV、分离温度为20 ℃时,D,L-丙氨酸和盐酸班布特罗的分离度均达到最大值,故确定两种分析物的最佳CEC运行条件为pH=8.5、20 mmol·L-1醋酸铵缓冲溶液、电压为20 kV、温度为20 ℃、50 mbar×3 s压力进样、紫外检测波长为200 nm。在最佳CEC分离条件下,D,L-丙氨酸和盐酸班布特罗均达到基线分离,分离图如图5和图6所示。利用本文自制的6-EA-β-CD电色谱整体柱,在最佳CEC运行条件下,盐酸班布特罗对映体在7 min内实现基线分离,与刘树仁等制备的整体柱对盐酸班布特罗对映体拆分结果[18]相比,两整体柱均能实现对盐酸班布特罗对映体的拆分,但本文的6-EA-β-CD电色谱整体柱能在更短时间内实现对盐酸班布特罗对映体的基线分离,更加高效快速。


图4 CEC运行条件对分离度的影响
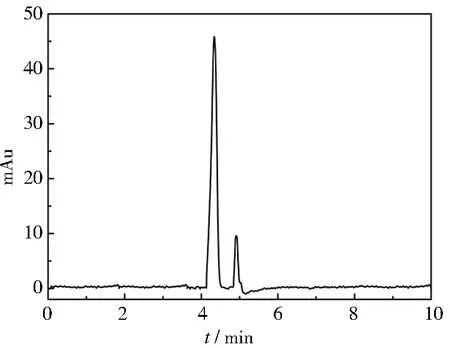
图5 丙氨酸电色谱分离图
3 结 论
采用简单的制备方法成功制备了一种新型毛细管电色谱整体柱,即6-EA-β-CD毛细管电色谱整体柱。在优化的CEC运行模式下,D,L-丙氨酸及盐酸班布特罗均在较短的时间内实现基线分离。该整体柱具有试剂消耗少和分离能力强的特点,为今后研究提供一定的基础。
猜你喜欢
高等学校化学学报(2024年2期)2024-03-06 06:31:12
好孩子画报(2020年2期)2020-03-30 03:47:05
广东医科大学学报(2020年6期)2020-02-06 06:00:38
知音海外版(上半月)(2018年5期)2018-05-23 03:11:20
中成药(2017年9期)2017-12-19 13:34:31
创新作文(小学版)(2017年22期)2017-04-04 02:17:16
癌变·畸变·突变(2016年5期)2016-08-22 05:55:18
畜牧兽医学报(2015年3期)2015-07-05 08:22:42
郑州大学学报(理学版)(2013年2期)2013-03-11 20:30:30
食品科学(2013年24期)2013-03-11 18:30:31
