
肠道功能障碍诱发帕金森病的机制*
2021-08-06 08:52:34郭轩彤陈祖昕路中华陈立铭王彦春刘欣安
华中科技大学学报(医学版) 2021年4期
郭轩彤, 陈祖昕, 曹 旭, 路中华, 陈立铭, 王彦春, 刘欣安△
1湖北中医药大学针灸骨伤学院,武汉 430065 2深港脑科学创新研究院,中国科学院深圳先进技术研究院/中国科学院深圳理工大学,脑认知与脑疾病研究所,深圳 518055 3深圳大学总医院神经内科,深圳 518055 4湖北中医药大学中医临床学院,武汉 430065
帕金森病(Parkinson’s disease,PD)是继阿尔兹海默症之后的全球第二大神经退行性病变,其典型的病理改变是错误折叠的α-突触核蛋白(α-synuclein,α-syn)不断聚集,以及大脑内黑质致密部多巴胺能神经元的变性与死亡,但其具体的致病机制尚不完全清楚。虽然越来越多的药物和非药物手段能改善部分PD运动障碍和非运动症状,但是仍无法逆转α-syn的不断聚集和多巴胺能神经元的持续性死亡,所以,进一步探究PD的发生发展机制对于更好地理解PD以及研究治疗方案都至关重要。大量的研究已经证实肠道菌群与神经元的发育、行为的调控以及胶质细胞的功能障碍均有密切的关系[1-3]。肠道功能紊乱或肠炎与PD之间的联系及其潜在的机制是近年来的研究热点。本文现就肠道功能障碍诱发PD的机制进行系统性回顾与分析,以利于PD脑肠轴机制及其临床应用的进一步研究。
1 肠道功能紊乱或增加PD发病风险
2003年Braak等[4]通过尸检观察到,α-syn起源于迷走神经的运动背核,并从此处发展到包括黑质的其他大脑区域,由此提出了PD病程的不同阶段学说。结合这一学说,以及特发帕金森病(idiopathic Parkinson’s disease,iPD)病例显示出的肠道神经系统和迷走神经背侧运动核受累的现象,Lionnet等[5]提出假说,大脑内聚集的路易小体很有可能来源于肠道,而连接大脑与肠道的迷走神经可能介导了α-syn的转运。临床已确诊PD患者的肠道中出现错误折叠的α-syn,由此得出PD源自于肠神经系统的理论假设。其后有诸多研究围绕着脑肠微生物轴机制开展,也有越来越多的证据表明PD的发生发展极有可能与肠道功能存在密切联系。
1.1 PD患者的胃肠道症状提示肠道功能紊乱可能诱导PD的发病
胃肠道症状是PD患者最常见的非运动症状之一,其症状具有多样性,并且涉及整个胃肠道,在PD的各个阶段均表现明显。超过30%的PD患者均有不同程度的胃肠道症状,包括恶心呕吐、饱腹感、胃排空延迟、胃轻瘫、腹胀等[6]。便秘,作为PD早期最常见的胃肠道症状之一,往往在PD确诊前15年已经出现。便秘在PD患者中的发生率为28%~80%,与同龄非PD患者相比,PD患者发生便秘的概率高达6倍及以上[7]。说明胃肠道症状的出现可能早于运动障碍,并且作为PD的高危因素存在,提示PD的发生发展极有可能与肠道功能密切相关。此理论为PD患者的病理生理研究提供了新的诊断思路,也为早期PD的外周诊断提供了新的可能依据。
1.2 PD的风险突变基因富亮氨酸重复激酶2(LRRK2)突变体与肠炎相关
除了PD患者的胃肠道前驱症状发生率较高这一突出特点之外,PD与炎症性肠病患者的肠道组织存在相同的致病危险基因——LRRK2突变体。LRRK2,是由人PARK8基因编码的一个酶。在PD病例中,90%发病原因不明,被称为iPD;剩下的10%具有较为明确的遗传基础,表现出家族发病趋势,其中LRRK2错义突变是常染色体显性遗传PD最常见的原因[8]。近期研究发现,无突变的iPD患者也存在LRRK2蛋白过度激活,神经元细胞自噬功能受损,导致α-syn在脑内的异常积累,参与PD的发展[9-10]。一项覆盖1.7亿人的调查研究发现,炎症性肠病患者比非炎症性肠病人群罹患PD的可能性高出28%。而LRRK2将两种看似不相关的疾病联系在一起的可能原因是:LRRK2是免疫系统中重要的调节因子,PD动物模型以及PD患者的大脑尸检均能测出炎症介质的升高,同样,全身炎症和自噬受损也被认为是炎症性肠病发病的重要原因[7]。虽然目前仍不明确PD与炎症性肠病之间的具体联系,但二者具有相同的风险因素,肠道LRRK2及其相关的分子通路可能成为PD潜在的治疗靶点。
2 PD的致病性蛋白可能源于肠道并通过迷走神经上传至脑
病理性α-syn来自于肠道的假说,在Braak提出之后,得到了越来越多的证据支持。Stokholm等[11]分析了来自39例PD患者的胃肠道组织,这些活体组织的采集时间点约在患者出现运动症状7年前。研究发现在这39例患者中,有22例患者的肠道神经系统中存在磷酸化的α-syn沉积,这一结果支持PD起源于胃肠道的假设。在动物模型上也有相关的研究证据,如最新的一项研究发现α-syn通过与Tau蛋白互作,能促进彼此从肠道至脑的传播,触发黑质多巴胺能神经元丢失[12]。这些研究均说明α-syn极有可能来自于胃肠道并且从肠道传播至脑。在病理性α-syn从肠道移行至脑的过程中,迷走神经和肠上皮屏障这两种解剖结构在传递病理性物质和未知病原体进入肠神经系统这一过程中发挥关键作用[4]。进一步地,有研究者使用糖探针来研究PD患者的肠道通透性并发现,PD患者的肠上皮屏障存在与肠炎患者相似的功能障碍[13]。此外,闭合蛋白(occludin),一种参与调控肠屏障通透性的紧密连接蛋白,在PD患者的结肠活检中被发现其表达改变[14]。这些结果提示PD患者的肠道屏障功能下降,可能参与肠道的病理性α-syn移行至肠神经元入脑的病理过程。
根据Braak的理论,迷走神经是病理性α-syn从肠道移行至脑的必要途径。这一理论在2019年得到了支持,Kim博士及其同事发现在小鼠十二指肠和幽门部注射的α-syn经迷走神经移行到黑质、蓝斑、嗅球、小脑等脑区聚集沉淀,并引起PD相关运动障碍及非运动症状[15]。由此表明,来自于肠道的α-syn通过迷走神经上行至迷走神经背侧运动核,然后在不同的脑区逐渐传播,最终引起PD。但是对于PD是否起源于肠道,目前仍有争议。《Brain》近期发表的一项以狒狒为模型的研究发现,α-syn不仅能从肠道传到大脑,也能进行反向传播,并提出体液循环而非迷走神经可能是其主要的传播途径[16]。所以,肠屏障功能障碍引起的病理性α-syn自肠道移行至脑而诱发PD,以及从肠神经系统到迷走神经系统的传播途径,需要更多的基础研究及临床研究证据加以证实。
3 PD患者的特征性粪便菌群改变参与PD发病的机制
尽管不存在两个人拥有完全相同的肠道菌群的情况,但是同一类患病人群的菌群可能表现出相似的特征性改变。已有报道显示PD患者肠道微生物组的组成特征是:阿克曼菌、双歧杆菌、肠杆菌及乳杆菌的丰度增加,属于拟杆菌门的丙酸杆菌大量减少[17]。PD患者肠道微生物群的组成变化会破坏其正常的稳态,引起肠道屏障功能障碍以及细菌的代谢产物的变化,从而诱发大量来自于肠道的内毒素——脂多糖(lipopolysaccharide,LPS)进入体内,激活巨噬细胞在内的免疫细胞活性,并激活肠道炎性小体释放大量的促炎因子,这些炎性因子又能触发更多免疫细胞活性,包括颅内的小胶质细胞,进一步释放更多的促炎因子,这一前馈级联反应能引起包括肠道在内的全身以及中枢神经系统的持续性炎症反应。
3.1 PD患者肠道菌群失衡
当肠道微生物群的稳态被破坏,大量的致病菌可引起肠道黏膜、大脑以及全身多系统功能障碍[18],例如炎症性肠病、肥胖/代谢综合征、PD以及其他多种慢性疾病[19]。对PD患者肠道菌群的研究,大多使用细菌16s核糖体RNA(rDNA扩增子)对不同可变区域进行测序,以鉴定粪便样本中的细菌谱,结果发现PD患者肠道中的菌群与正常同龄人差异明显[20-21]。2015年的一项研究报告指出,PD患者的粪便样本中,与抗炎效应相关的细菌减少,如劳特菌(Blautia)、粪球菌(Coprococcus)、罗斯拜瑞菌(Roseburia),且在PD患者的肠道黏膜中发现粪杆菌属(Faecalibacterium)减少,劳尔菌属(Ralstonia)增加,促使结肠内的微生物平衡转变为炎症倾向表型[22]。另一项研究表明,PD患者粪便中乳酸杆菌的数量较高,而普雷沃菌属(Prevotella)、酪酸菌属(Clostridium)和脆弱拟杆菌(Bacteroidesfragilis)的数量较少,且普雷沃菌属(Prevotella)数量的减少可能导致粘蛋白合成减少,肠道通透性增加[23]。这种微生物失调的状态可引起肠黏膜的不完整以及肠道屏障功能障碍,直接导致血液中的内毒素(例如LPS,一种可以诱导PD动物模型的细菌代谢产物[24])水平升高[24],进而引起中枢神经系统的炎性反应[25],促进病理性α-syn的聚集[26],而这种病理性α-syn的聚集又能刺激胶质细胞的进一步活化,触发中枢神经系统持续性炎症反应[27],最终出现PD相关的运动及非运动症状。PD患者肠道菌群的变化可能引起肠黏膜的不完整以及肠道屏障功能障碍,这种前馈级联反应在PD的发展过程中至关重要。
3.2 肠道微生物代谢产物异常
细菌的代谢产物包括能激活小胶质细胞触发炎症反应的促炎产物如LPS等[23-24],以丁酸盐为代表的短链脂肪酸(short chain fatty acid,SCFA)等抗炎产物[28],肠肽类[包括神经递质,如γ-氨基丁酸(γ-aminobutyric acid,GABA)、血清素(serotonin)、多巴胺(dopamine)等[29],和神经调质,如胃肠道细胞产生的激素[30]等]。细菌代谢产物的变化能通过多种途径诱发神经退行性疾病,其中主要包括以下几条通路。
3.2.1 异常的肠道微生物代谢产物 影响肠道屏障功能主要由肠道微生物产生的2种物质介导:细菌的成分(如LPS)和细菌的代谢产物(如SCFA)。两者均能影响肠道屏障的完整性,而这种完整性在肠道、脑和全身免疫系统中都有举足轻重的作用[31]。具体来说,SCFA的减少能触发肠道屏障功能障碍和肠道炎症反应,最终导致神经炎症以及神经变性[32]。在PD患者肠道内,这种SCFA减少所引起的肠道屏障功能障碍和内毒素大量产生的病理表现都非常显著[31]。临床确诊的PD患者与同龄对照组相比存在明显的肠道屏障功能障碍,这是由于PD患者的血清LPS结合蛋白LBP与LPS结合诱导肠道免疫反应;诱发肠道致密连接蛋白表达异常[14,33]。此外,PD患者α-syn在结肠黏膜中的聚集也导致肠道屏障功能障碍[33]。以上研究结果均表明:肠道微生物代谢产物的失调能通过诱导肠道屏障功能障碍,进而诱发持续性的中枢神经系统炎症反应,从而在PD的发生发展过程中扮演着重要的角色[31-32]。
3.2.2 炎性小体激活机制 炎性小体作为天然免疫系统的重要组成部分,在神经退行性疾病、肠炎、癌症、2型糖尿病等多种疾病中都发挥着举足轻重的作用[28]。炎性小体是一个蛋白复合物,主要包含受体蛋白(receptor)、接头蛋白(adaptor)、ASC蛋白组分(apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain)以及下游的效应蛋白Caspase-1。当受体蛋白被激活后,会随之吸引ASC与Caspase-1聚集组装成炎性小体,从而诱导Caspase-1自切割并活化。活化的Caspase-1一方面能促进包括白介素-1β(IL-1β)和白介素-18(IL-18)在内的多种促炎细胞因子的成熟和分泌;另一方面能引发细胞焦亡(pyroptosis)清理病原体和受损细胞。绝大部分炎性小体的受体蛋白是通过直接或间接结合一种(或少数几种)特异的激动剂被激活的,但NLRP3(NLR family pyrin domain containing 3)作为NLR受体蛋白家族成员是一个例外,它能够被来源、化学组成、结构性质均不同的激动剂所激活。在PD的发生发展过程中,NLRP3扮演着重要角色。肠道屏障功能障碍引起的内毒素血症(即血液中流动的LPS)能被Toll样受体(Toll like receptors,TLRs)识别并激活,活化的TLRs一方面能激活NLRP3炎性小体[28,34]并释放促炎因子[35],尤其是白介素-1β(IL-1β)、白介素-1α(IL-1α)、白介素-18(IL-18)以及白介素-33(IL-33)[36]。这些促炎因子除了能刺激以巨噬细胞为主的外周炎症反应以外,还能刺激胶质细胞介导的中枢神经系统炎性反应[34]。另一方面,TLR的激活能诱导线粒体功能障碍[37],并促进病理性α-syn聚集[38],而这又能进一步诱导NLRP3的激活以及募集更多促炎因子的释放,产生中枢神经系统持续的促炎/氧化应激反应,导致更多的病理性α-syn聚集,最终导致多巴胺能神经元变性与丢失以及PD相关症状的发生发展。最近一项对PD患者的尸检研究表明,NLRP3炎性小体在PD患者脑内明显上调且大概率与小胶质细胞共定位[28]。在注射α-syn原纤维、MPTP或鱼藤酮的PD动物模型中,抑制NLRP3活性对各种因素诱导的神经变性均存在明显的保护作用[28,39-40]。这些研究均证实了NLRP3炎性小体在PD发展过程中的重要性。
3.2.3 肠肽和糖异生机制 细菌代谢产物的变化能阻碍肠肽和脑源性神经营养因子(brain derived neurotrophic factor,BDNF)的产生。短链脂肪酸(SCFA)和次级胆汁酸作为肠道细菌的代谢产物,都能促进胰高血糖素样肽-1(glucagon like peptide 1,GLP-1)和葡萄糖依赖性促胰岛素多肽(glucose dependent insulinotropic polypeptide,GIP)的产生[41]。一方面,GLP-1能通过纠正胰岛素抵抗来改善线粒体功能,减少超氧化物歧化酶的产生,发挥保护神经元正常功能的作用[42]。GLP-1受体的激活还能抑制NLRP3炎性小体的活性[43],可能通过产生胶质细胞源性的神经营养因子(glial derived neurotrophic factor,GDNF)和BDNF来抑制神经元变性及神经炎性反应,对神经系统起保护作用[44]。另一方面,GLP-1能促进BDNF的产生,这种神经营养因子对于维持黑质多巴胺能神经元至关重要[45]。
除肠肽以外,肠道糖异生也是影响中枢神经系统炎症和神经退行性病变的另一个重要机制。PD患者的肠道微生物异常,由此产生的SCFA减少。肠道糖异生-迷走神经-BDNF是正常脑葡萄糖代谢的重要机制[46]。最近的研究表明,SCFA如丙酸丁酸脂可以通过刺激肠上皮细胞中的肠道糖异生来调节宿主代谢,主要是通过肠道糖异生-迷走神经-BDNF信号通路和调节GLP-1/GIP表达来纠正胰岛素抵抗,防止肠道屏障功能障碍和NLRP3激活。一方面,SCFA(尤其丁酸和丙酸)能通过刺激肠上皮细胞糖异生以及刺激GLP-1和GIP的表达增高来维持肠壁的完整性,减少NLRP3的激活[18,41],从而减轻中枢神经系统的炎症反应。另一方面,肠道糖异生也可以通过迷走神经影响脑内BDNF的产生,而BDNF能维持神经细胞正常功能和脑内正常的胰岛素信号[47]。PD患者脑内葡萄糖代谢异常与低水平的BDNF有关,而这种低水平的BDNF又与肠道糖异生密切相关,说明由微生物所引起的肠道糖异生通路异常可能通过葡萄糖异常代谢的方式参与PD的发生发展。
综上,PD患者特征性的肠道微生物失调一方面可触发肠道屏障功能障碍,诱发肠道炎症从而促使病理性α-syn从肠道移行至大脑,参与PD的发生发展;另一方面,肠道微生物变化使得细菌的代谢产物变化,诱导炎症反应通过体液循环途径参与PD的发生发展过程(图1)。
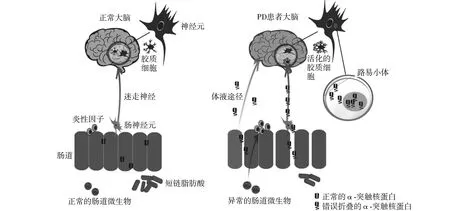
脑-肠-微生物轴的功能失调在PD发病中起重要作用,肠道微生物组的改变增加了PD的患病风险,与微生物失衡、短链脂肪酸、LPS以及肠道屏障功能的改变相关。
4 调控胃肠道功能治疗PD的新策略及研究展望
阐明肠道菌群及功能改变与PD发病机制之间的时态和相互关系,这一前沿领域的工作进展对PD的治疗提供了新的策略和思路,具有重要的临床意义。更好地理解脑-肠道-微生物轴相互作用将促进对PD的病理生理学及其临床诊疗的深入认识,如通过肠神经系统内的PD相关疾病标志物对PD进行极早期诊断,以及提示新的治疗手段等。对PD患者或PD高风险人群实行针对改变肠道微生物群组成及提高肠道上皮屏障完整性的饮食或药物干预,可能对PD的神经退行性变的病程有所延缓或逆转。通过饮食改变肠道微生物组成,进而影响脑肠轴功能。不同的营养干预措施包括益生菌的使用致力于使疾病状态下异常肠道微生物组正常化,进而改善神经系统疾病的症状,延缓疾病进展。
目前的研究证明,使用益生菌能改善PD患者大便硬度和排便习惯[48],也有研究表明益生菌的使用可以降低PD患者MDS-UPDRS评分[49],但目前的研究尚不能确定益生菌治疗是否能减缓PD的进展。粪便移植作为一种新兴的治疗方式,被证实对MPTP处理的PD老鼠具有神经保护作用[50],但是粪便移植对PD患者的长期疗效和潜在不良反应尚无明确的报道加以证实。微生物疗法可能对患有胃肠道疾病的PD患者有益。但目前的研究数据有限,且多集中于动物实验,临床实验相对匮乏,益生菌的治疗作用不应被盲目夸大,因为外源性益生菌对PD患者的自身稳定菌群和肠道微环境的长期影响尚不清楚。因此益生菌尚不能成为PD患者胃肠道治疗的一线用药。人的肠道菌群组成和多样性,以及PD的发病机制都存在个体差异,因此未来对PD的微生物治疗应朝着个性化精准治疗的方向发展,并需要更多关于肠道微生物参与PD发生发展的基础和临床研究,从而为PD的临床治疗提供新的方案。
本综述中讨论的PD的脑-肠-微生物菌群的相互作用的神经环路及其分子机制(图1),提示在PD的临床诊断中应考虑肠道微生物的特征性改变,并将为益生菌、粪菌移植、肠道微生物群的修饰等肠道纳米药物在PD的预防、治疗或作为辅助治疗的潜在用途提供依据,这也将进一步促进生物学、化学和材料学等多学科交叉发展,实现肠道菌群的精准调控,从而推动PD临床治疗的新方法。
猜你喜欢
云南中医中药杂志(2023年12期)2023-12-22 06:19:04
江苏安全生产(2022年8期)2022-11-01 09:14:32
小资CHIC!ELEGANCE(2021年36期)2021-10-15 14:36:34
湖南中医药大学学报(2021年8期)2021-09-22 20:00:22
艺术评鉴(2020年5期)2020-04-30 06:47:57
医学综述(2020年18期)2020-02-16 20:47:55
人大建设(2018年10期)2018-12-07 01:13:46
中成药(2018年5期)2018-06-06 03:12:06
中国组织化学与细胞化学杂志(2017年1期)2017-06-15 20:27:43
西南军医(2015年3期)2015-04-23 07:28:32
