
糖异生相关通路的研究进展及中药的改善作用*
2023-12-22 06:19:04胡文心
云南中医中药杂志 2023年12期
胡文心,杨 杰
(中国药科大学,江苏 南京 211198)
2型糖尿病(T2D)是由于胰岛素抵抗和(或)胰岛素分泌缺陷导致的胰岛素信号传导缺乏引起的慢性高血糖症。据估计,2017年全球有4.51亿(18-99岁)糖尿病患者,预计到2045年将增加到6.93亿[1]。T2D的患病率和发病率占所有糖尿病病例90%以上[2],可引起多器官损伤和多种并发症,对人类生命健康与安全构成了巨大威胁。因而,控制血糖水平,延缓T2D进程不可忽视。
肝脏是葡萄糖产生的主要部位,对于维持正常的葡萄糖稳态至关重要,它在禁食期间产生葡萄糖并在餐后储存葡萄糖。然而,这些过程在T2D中失调,这种不平衡导致禁食和餐后状态下的高血糖[3]。胰岛素降低餐后血糖,而空腹血糖的稳态主要由胰高血糖素作用下的肝糖异生维持。肝胰高血糖素紊乱所致的肝糖异生异常是糖尿病中空腹血糖升高的直接原因[4]。可见确定调节肝脏糖异生的分子机制对T2D治疗策略的改进至关重要。中药具有多组分、作用多靶点的特点,在调节复杂疾病系统如T2D占有优势,不易产生耐药性[5]。因此,本文还针对近年来中药改善糖异生的作用研究进行了汇总。
1 肝内糖异生途径及主要调节激素
1.1 肝糖异生途径 糖异生是指将一些非碳水化合物底物(如丙酮酸、乳酸、甘油、生糖氨基酸等)转化为糖的过程。在哺乳动物中,肝脏是糖异生的主要器官。如图1所示,
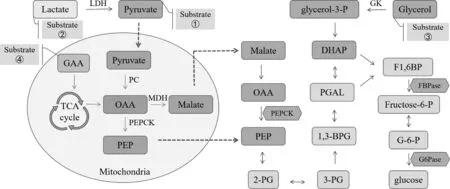
图1 肝糖异生途径示意图
丙酮酸(pyruvate)或乳酸(lactate)被乳酸脱氢酶(LDH)催化为丙酮酸后进入线粒体,在线粒体丙酮酸羧化酶(PC)作用下转化为草酰乙酸(OAA),然后被磷酸烯醇式丙酮酸羧激酶(PEPCK)催化为磷酸烯醇式丙酮酸(PEP),进入细胞质进行糖异生循环。OAA也可以直接在线粒体内被还原为苹果酸(Malate)穿梭到细胞质中,再被丙酮酸脱氢酶(MDH)氧化为OAA,通过胞质PEPCK磷酸化为PEP。PEP在烯醇化酶催化下转变为2-磷酸甘油酸(2-PG),2-PG重排生成3-磷酸甘油酸(3-PG),再转变为3-磷酸甘油醛(PGAL),PGAL可异构为磷酸二羟丙酮(DHAP),其接着转化为果糖-1,6-二磷酸(F1,6BP),在果糖-1,6-二磷酸酶(FBPase)作用下形成果糖-6-磷酸(Fructose-6-P),然后通过磷酸葡萄糖异构酶转化为葡萄糖-6-磷酸(G6P)。最后,G6P通过葡萄糖-6-磷酸酶(G6Pase)转化为葡萄糖。甘油经血液循环进入肝脏后被甘油激酶(GK)转化为3-磷酸甘油(glycerol-3-P),其转化为DHAP进入糖异生途径。生糖氨基酸可转化为参与三羧酸循环(TCA cycle)的中间物,比如天冬氨酸可生成OAA进入糖异生途径。糖异生可以说是糖酵解的逆向过程,但其中有三个步骤不可逆,PEPCK、FBPase、G6Pase是这三步关键的限速酶[6]。
1.2 主要调节激素
1.2.1 胰高血糖素 胰高血糖素(glucagon)是胰腺α细胞分泌的多肽激素,是一种关键的糖异生调节因子。空腹时胰岛α细胞感受周围环境低血糖的变化释放胰高血糖素,通过肝糖异生和输出保持空腹血糖稳定。胰高血糖素与肝细胞膜上G蛋白受体结合激活肝细胞内环磷酸腺苷/蛋白激酶A(cAMP/PKA)信号系统,促进PKA介导的cAMP反应元件结合蛋白(CREB)磷酸化和盐诱导激酶2(SIK2)的抑制,导致cAMP调控的转录共激活因子2(CRTC2)去磷酸化,核易位和募集到CREB占据的基因,调控糖异生限速酶基因的转录(G6pase、PEPCK)[7],促进肝糖异生和输出(图2)。研究发现胰高血糖素分泌异常的人群更易发展为糖尿病[8]。虽然临床讨论通常集中在胰岛素的作用上,但胰高血糖素在T2D研究上同样重要[9]。
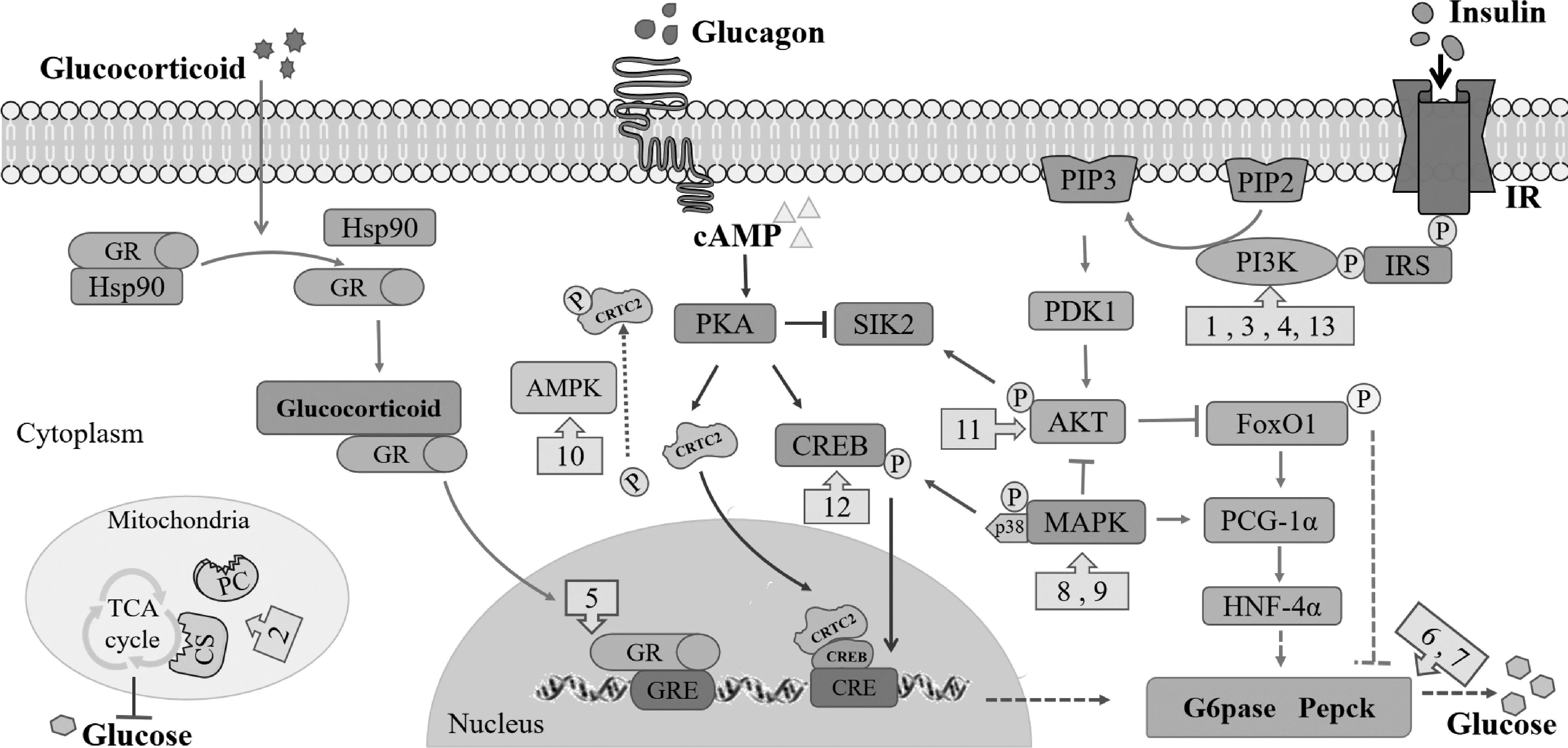
图2 糖异生相关作用通路示意图
1.2.2 胰岛素 胰岛素(Insulin)是胰腺β细胞分泌的多肽激素,餐后胰高血糖素水平下降,胰岛素浓度升高,通过刺激肝脏葡萄糖摄取和糖原储存以及抑制肝脏葡萄糖产生(HGP),维持人体内正常葡萄糖水平。胰岛素通过与跨膜胰岛素受体的结合触发磷脂酰肌醇-3-激酶(PI3K)-蛋白激酶B(AKT)-Forkhead转录因子(FoxO1)途径抑制糖异生[10],AKT磷酸化可以激活SIK2作用于糖异生信号[7]。大多数人体细胞利用葡萄糖作为主要底物,细胞摄取需要胰岛素。因此,胰岛素信号传导对这些组织至关重要。然而,由于各种分子途径的破坏而导致胰岛素敏感性降低会导致胰岛素抵抗[11]。胰岛素抵抗是T2D的典型特征,期间,胰岛素不能刺激肝糖摄取,未能抑制糖原分解和糖异生,餐后HGP异常升高[12]。
1.2.3 糖皮质激素 糖皮质激素(glucocorticoid)是由肾上腺分泌的甾体激素,在调节哺乳动物的葡萄糖稳态中起着关键作用。在肝脏中,糖皮质激素可以促进糖异生。GC主要通过细胞内受体糖皮质激素受体(GR)传递信号。在与GC结合之前,GR存在于细胞质中并与Hsp90伴侣复合物相关。与GC结合后,GR从Hsp90伴侣复合物中解离并进入细胞核,其中GR被募集到特定的基因组序列中,称为糖皮质激素反应元件(GRE),已经证明其能启动G6pase、PEPCK的转录调节[13](图2)。研究发现TAZ通过抑制GR从而调节糖异生[14]。
2 糖异生相关调控通路
2.1 CRTC2/CREB CREB是糖异生基因表达的关键调节因子,CREB的转录活性受辅助激活因子CRTC2所调节。在基础状态下,CRTC2以磷酸化的形式隔离在细胞质中,当胰高血糖素通过cAMP/PKA信号激活CREB时,CRTC2随即入核和CREB 的bZIP结构域结合,稳定CREB 和靶基因启动子的结合,转录调节糖异生限速酶的表达,促进糖异生[7](图2)。这一信号的专属性响应表明了CRTC2在糖异生调节中的不可或缺性。有研究发现蜂胶通过破坏CREB/CRTC2转录复合物的形成来降低肥胖小鼠的空腹血糖水平[15],Sam68通过与CREB/CRTC2复合物中的CRTC2相互作用并占据启动子的CRE基序来增加CREB/CRTC2的交互性,导致糖异生基因表达和葡萄糖产生[16]。已经证明,腺苷酸活化蛋白激酶(AMPK)可介导CRTC2磷酸化减少其入核改善肝糖异生[7]。
2.2 PI3K/AKT/FoxO1 PEPCK和G6Pase是FoxO1的重要靶基因,催化糖异生中的限速步骤。胰岛素受体底物(IRS)通过与磷脂酰肌醇-3-激酶(PI3K)结合并激活它来触发随后的信号转导,催化磷脂酰肌醇-4,5-二磷酸(PIP2)转化为磷脂酰肌醇3,4,5-三磷酸肌醇(PIP3)。PIP3产生激活丙酮酸脱氢酶激酶1(PDK1),导致随后AKT被PDK1上的Thr308磷酸化,然后AKT通过磷酸化和抑制FoxO1来抑制糖异生基因表达[10](图2)。有研究发现葛根素通过PI3K/AKT 介导FoxO1磷酸化以及PEPCK和G6Pase的下调减少糖异生[17]。丝裂原活化蛋白激酶(MAPK)途径的过度激活会导致AKT磷酸化受损[18]。
2.3 PGC-1α/HNF-4α 肝细胞核因子4α(HNF4α)是肝细胞中表达的核受体,并调节参与糖脂代谢的基因的表达,包括控制糖异生的基因。过氧化物酶体增殖受体γ辅激活因子α(PGC-1α)是肝脏糖异生的关键调节因子,HNF4α是PGC-1α介导的糖异生所必需的,通过共激活PEPCK和G6Pase,是与PGC-1α有关的重要肝脏富集成分,PGC-1α还受FoxO1激活刺激糖异生(图2)。在HNF4α缺失后的所有喂养条件下,PEPCK和G4Pase表达均下降[19]。研究表明,PGC-1α通过共激活HNF4α和FoxO1促进肝糖异生[20]。
3 中药改善糖异生的相关研究进展
肝糖异生异常增强是T2D发病的重要机制,近年来关于中药在糖异生中作用得到了广泛的关注,表1对近年来中药复方、单味药以及中药单体相关作用机制研究进行了总结,并在图2中注明,图2中编号1-14分别对应表1中不同中药的相关作用通路,希望能为临床上治疗T2D提供新的思路。
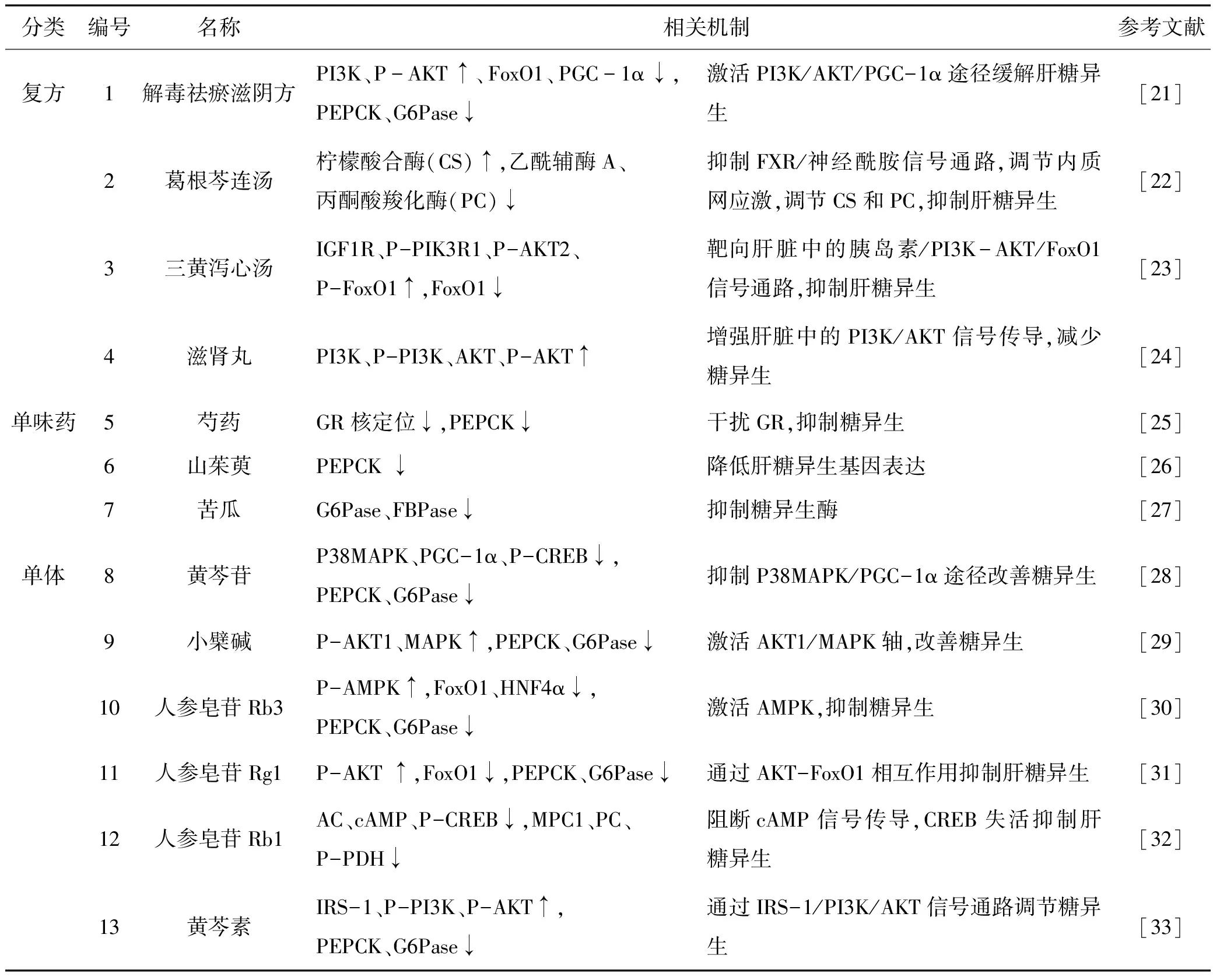
表1 中药改善糖异生的相关作用机制
4 小结
当前,T2D患病率呈持续增长趋势,是人类健康的重大隐患。临床上一线药物二甲双胍具有良好的疗效,但是随之出现的不良反应限制了其使用。中药具有副作用较少、多靶点整体调节的特点,在T2D治疗中具有良好的应用前景。肝糖异生异常是T2D中空腹血糖升高的重要原因,其调节在T2D防治上不容忽视。本文综述了目前常见的糖异生调控激素靶点及近年来中药改善糖异生的相关作用通路,希望能为新药研发提供潜在靶点。近年来关于中药改善糖异生的作用研究取得了一定的进展,但是许多研究基本上是基于动物与细胞水平进行,还需更广泛的临床研究加以验证。同时,大多数研究都是基于单一靶点或调节通路上,靶点与调节通路之间的联系还需开展更为深入的研究。
猜你喜欢
工业微生物(2024年1期)2024-02-29 07:36:50
生物加工过程(2023年6期)2023-12-11 03:27:52
牡丹江医学院学报(2021年5期)2021-12-05 08:01:51
湖南中医药大学学报(2021年8期)2021-09-22 20:00:22
肉类研究(2021年5期)2021-07-11 16:10:36
医学综述(2020年18期)2020-02-16 20:47:55
国际呼吸杂志(2019年22期)2019-12-09 09:20:38
中成药(2018年5期)2018-06-06 03:12:06
中国卫生标准管理(2015年18期)2016-01-20 09:27:07
成都医学院学报(2015年3期)2015-12-06 02:31:38
