
TREX1基因变异致Aicardi-Goutières综合征1例报告
2021-07-16 12:03高向莹杨学梅
临床儿科杂志 2021年7期
高向莹 杨学梅 陈 虹
1.兰州大学第一临床医学院(甘肃兰州 730000);2.兰州大学第一医院(甘肃兰州 730000)
Jean Aicardi和Francoise Goutières于1984年报道一种家族性脑病,其后被命名为Aicardi-Goutières综合征(Aicardi-Goutières syndrome,AGS)。AGS是一种罕见的单基因遗传性脑病,多为常染色体隐性遗传,极少数为常染色体显性遗传。AGS 与颅内钙化、白质异常和脑萎缩等影像学改变有关,还与脑脊液中淋巴细胞增多和高水平的干扰素有关。高水平的干扰素主要损害神经系统,包括易激惹、肌张力减退、癫痫、进行性小头畸形、智力及运动发育迟缓、瘫痪等,还可表现出多种神经外症状,包括皮肤红斑及冻疮样改变、先天性青光眼、自身抗体水平升高、甲状腺功能减低、溶血性贫血、脱髓鞘周围神经病、系统性红斑狼疮等[1]。迄今为止国际报道的AGS致病基因有7种,包括TREX1、RNASE2B、RNASE2C、RNASE2A、SAMHD 1、ADAR和1 F 1 H 1。查阅相关文献,国外报道超过200例AGS,日本报道AGS发病率约为1/900万[2]。查阅国内数据库[3],尚无AGS患病率的相关报道,报道7例AGS,其中与TREX1基因变异相关病例3例,RNASE2A基因、ADAR基因、1F1H1基因变异各1例,1例未行基因检测;年龄最小者5月龄,最大者39岁。目前国内尚无新生儿AGS的相关报道。本文回顾分析近期收治的1例新生儿期确诊为AGS患儿的临床及家系基因特征。
1 临床资料
患儿,女,系G2P2,40+1周顺产。患儿出生时羊水Ⅱ°污染,生后全身皮肤青紫,自主呼吸弱,四肢肌张力低,Apgar评分1分钟7分,5分钟8分,出生体质量 2 400 g,余无特殊。患儿出生后以新生儿肺炎转入当地医院新生儿监护室,生后第2天即出现发热、喂养困难,伴有呛奶、呼吸急促;查血常规白细胞17.75×109/L,中性粒细胞0.67,淋巴细胞0.26,单核细胞0.61,C反应蛋白11.76 mg/L;脑脊液无异常;诊断为新生儿败血症,给予抗感染治疗;治疗10余天仍有间断发热,呼吸急促加重,遂转至兰州大学第一医院住院治疗。入院诊断为:新生儿败血症、新生儿肺炎。父母非近亲婚配,无相关遗传病史。同母异父的哥哥体健。
入院体格检查:神志清,反应可,哭声有力,呼吸急促;口唇发绀,全身皮肤苍白,无白斑及冻疮样改变;特殊面容,头型尖、头围30 cm、眼距宽、眼裂窄、鼻梁低平、上唇短小、短下颌;心肺腹查体无异常;四肢肌张力低;觅食反射、吸吮反射、拥抱反射、握持反射正常引出。入院实验室检查:血、尿、粪常规无异常;肝、肾、心肌酶、离子、血糖无异常;凝血功能及甲状腺功能无异常;神经元特异性烯醇化酶40.31 ng/mL;免疫球蛋白及T 细胞亚群无异常;TORCH、13 种呼吸道病原核酸检测均阴性;尿巨细胞病毒检测阴性;GM 试验及G 试验阴性。痰细菌培养:分别培养出大肠埃希菌、肺炎克雷伯杆菌生长。脑脊液常规:白细胞31×106/L,单核细胞0.50,中性粒细胞0.10,淋巴细胞0.40;脑脊液蛋白1.52 g/L,糖和氯化物无异常;脑脊液细菌学检查无异常。遗传代谢筛查:血氨基酸、肉碱、琥珀酸丙酮无显著异常;尿液有机酸分析无显著异常。心脏彩超示动脉导管未闭,卵圆孔未闭。头颅CT 示双侧基底节区及侧脑室旁斑片样高密度影双侧额叶斑片状密度稍高影大枕大池(图1)。患儿28日龄时头颅磁共振成像(MRI)示双侧基底节区及侧脑室旁多发斑点状短T1短T2信号影;基底动脉走形迂曲;双侧丘脑区乳酸峰略升高,考虑缺氧改变(图2A)。38日龄时复查头颅MRI 示双侧基底节区及侧脑室旁多发斑点样异常信号范围较前略有缩小(图2B)。

图1 患儿头颅CT 表现
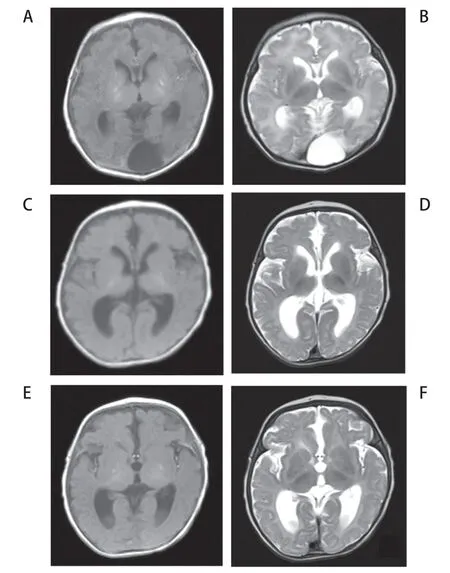
图2 患儿头颅MRI 表现
结合患儿临床表现及体征,不排除合并遗传、代谢性疾病。经医学伦理审核并征得患儿父母同意后,采集患儿及其父母外周血标本,送至北京智因东方转化医学研究中心进行全外显子测序,与人类2 万个基因组的全外显子区进行比对,对目标序列进行PCR 检测后,经ABI 3730 测序仪进行Sanger 测序验证,分析OMIM、MedGen等数据库已报道的疾病。结果显示患儿TREX 1基因发现2 个变异位点:c.457_c.458insA及c.517C>G,分别来自于父亲及母亲,见图3。TREX1基因c.457_c.458insA及c.517C>G变异位点的致病性尚未见文献报道。变异预测结果显示,上述2处变异位点均为致病可能性较高的基因变异[4]。本例患儿为杂合子,其父母均为表型正常的携带者,符合常染色体隐性遗传发病机制。结合患儿的临床表现、体征、影像学及基因检测,确诊为AGS1型。
患儿随访至3个月28天,头围31 cm、小下颌,生长发育迟缓(身长52 cm,体质量4 300 g),有肝大、肝酶增高等;复查头颅MRI示双侧苍白球见对称性点状分布短T 1 信号影;双侧内外囊及皮质脊髓束脑桥段T2信号增高;脑萎缩改变(图2C)。
2 讨论
AGS是由基因变异引起的遗传免疫介导的疾病,遗传方式为常染色体隐性遗传,少数为常染色体显性遗传,多认为与基因变异所致的Ⅰ型干扰素代谢异常有关,Ⅰ型干扰素水平增加可促进自身抗体的产生,最终导致全身性自身免疫损害[5]。AGS典型的临床表现为神经功能障碍,包括进行性小头畸形、痉挛、精神运动迟缓等,经典标志为颅内钙化、脑白质发育异常、脑萎缩及脑脊液中淋巴细胞百分比增加。
欧洲对110 例AGS 患者的研究表明,90.9%患者存在脑钙化,91.8%存在脑萎缩,99.2%患有脑白质病,27.7%有髓鞘变性[6]。AGS分为7型,分别为:TREX1-AGS1型、RNASEH2B-AGS2型、RNASEH2CAGS3型、RNASEH2A-AGS4型、SAMHD1-AGS5型、ADAR-AGS 6 型和IFIH 1-AGS 7 型[7],其中TREX 1-AGS1型最早发现,IFIH1-AGS7型最晚发现。AGS根据发病年龄分为早发型和晚发型,TREX1基因变异导致的AGS 多为新生儿期或婴儿期起病,称为早发型。早发型多表现为易激惹、喂养困难、小头畸形、异常动作、癫痫、骨髓抑制和肝功能异常。随后可出现神经系统退行性改变,典型的AGS影像学特征为弥漫性白质异常、颅内钙化及脑萎缩[8]。RNASEH2B基因变异所致的AGS多发生在一段正常发育期以后,通常为4月龄左右,称为晚发型。晚发型病例多为亚急性起病,其特征为极度易激惹、间歇性发热、头部生长缓慢、运动及智能的退行性改变,这种表现一般持续数月,病情进展缓慢。AGS的神经系统症状会在发病后逐步体现出来,如肢体痉挛、姿势异常、智力降低及头部控制不良,约50%患有癫痫。AGS的各个基因型都有可能出现冻疮样表现,特别见于脚趾和手指,也可见于外耳,冬天显著[9]。本例患儿于新生儿期起病,生后2天即出现发热、喂养困难,起初诊断为新生儿败血症,同期发现患儿有发育落后、小头畸形、短下颌、上唇短小、肌张力低等表现,头颅影像学检查示AGS 典型影像学表现。本例患儿与早发型AGS表现相一致。目前随访近4个月,存在生长发育迟缓及肝酶增高等表现,头颅MRI示进行性脑萎缩,目前缺乏典型皮肤表现及癫痫发作;基因检测示患儿携带2处TREX1基因变异位点,为AGS1型。
AGS 为一种单基因遗传病,目前与AGS 相关的变异基因有TREX 1、RNASEH 2 A、RNASEH 2 B、RNASEH 2 C、SAMHD 1、ADAR和IFIH 1[7]。对欧洲374 例AGS 患者的7 个基因变异类型及临床特征进行分析发现,22%有TREX 1基因变异,其次为RNASEH 2 B(36%)、RNASEH 2 C(12%)、SAMHD 1(13%)、RNASEH2A(5%)、ADAR(6%)和IFIH1(3%);11.4%的患者于生后即出现异常神经症状,68.6%在生后1 岁内出现神经功能障碍,18%发育无异 常[10]。本例患儿TREX1基因发现2个变异位点c.457_c.458insA及c.517C>G,分别来自于父亲及母亲,其父母均为表型正常的携带者,推测为常染色体隐性遗传。TREX 1基因编码一种具有3’外切酶活性的核蛋白,在DNA修复中起主要作用,并在控制自身免疫性疾病中具有重要作用[8]。与TREX1变异相关的人类疾病包括AGS、家族性冻疮狼疮、系统性红斑狼疮和视网膜血管病变合并脑白质营养不良。几乎所有TREX1变异的AGS 患者都存在严重的神经功能障碍,表现为痉挛、肌张力障碍、癫痫、皮质性失明和小头畸形,冻疮样改变的发生率为36.7%[11],大约2%的系统性红斑狼疮患者有TREX 1变异。颅内大血管病变多与SAMHD1变异有关,近期有报道1例TREX1基因变异所致颅内大血管病变[8]。本例患儿为TREX1基因变异,临床表型符合AGS,尚缺乏皮肤冻疮样改变及脑脊液淋巴细水平增多等典型表现,无癫痫发作,需在后期的随访中关注有无典型皮肤改变及癫痫发作。
AGS主要的影像学改变为颅内钙化、脑白质异常及脑萎缩。脑钙化可在75%~100%的患者中发现,钙化多分布于基底节区、侧脑室及双侧半卵圆中心,以双侧为主。钙化的数量、大小和形态不能决定病变严重程度,可随着时间的推移趋于稳定[5]。AGS 的另一个典型表现为白质营养不良,多发生于脑室周围,也可在额叶及颞叶区域,有时表现为囊性变。AGS 的第3个影像学特征是脑萎缩,尤其是脑室周围区域,有些还表现为脑沟增大、脑干和小脑萎缩及胼胝体发育异常[5]。脑脊液中淋巴细胞水平增多是AGS的重要实验室特征,但是,AGS 患者脑脊液中淋巴细胞在发病后早期都可降至正常水平。因此,脑脊液淋巴细胞数正常并不能排除AGS[12]。脑脊液中高水平的α-干扰素是AGS的可靠标志物,然而,很多医疗单位无法检测脑脊液α-干扰素。还有报道CXC趋化因子配体10在AGS患者的脑脊液中也显著升高,提示可能是AGS的另一种脑脊液标记物[13]。
目前,尚无针对AGS 的特异性治疗方法,多为对症治疗,包括控制癫痫、预防并发症、治疗肺部感染及饮食监测。AGS 存在α-干扰素代谢异常,抗干扰素α 抗体治疗的临床试验已在进行中。AGS 中逆转录酶抑制剂,如环状GMP-AMP合酶(circular GMPAMP synthase,cGAS)是自身免疫性疾病的关键驱动因素,cGAS 抑制剂可能是AGS 和自身免疫相关疾病的有效疗法,但尚未应用于临床中。静脉用人免疫球蛋白、激素及免疫抑制剂可改善AGS的神经系统退化表现,但仍待进一步研究[14]。AGS 生存质量差,预后不良,死亡率高,多遗留不同的神经系统后遗症,很难存活到成年。TREX1基因变异的AGS病死率最高,RNASEH2B变异的病死率低[1]。
猜你喜欢
中西医结合心脑血管病杂志(2022年14期)2022-08-19
体育科技文献通报(2022年3期)2022-05-23
保健与生活(2022年9期)2022-05-06
健康博览(2021年7期)2021-08-16
智慧健康(2021年33期)2021-03-16
保健与生活(2020年11期)2020-06-23
医学新知(2019年4期)2020-01-02
保健与生活(2019年15期)2019-09-12
家庭科学·新健康(2016年9期)2016-10-25
中国医药科学(2014年24期)2015-03-16
