
ALG1基因相关先天性糖基化障碍伴婴儿痉挛症2例报告及文献复习
2021-07-16 12:03王秋红邹丽萍王杨阳沈雁文石秀玉
临床儿科杂志 2021年7期
王秋红 邹丽萍 王杨阳 沈雁文 石秀玉
中国人民解放军总医院第一医学中心儿内科(北京 100853);北京脑重大疾病研究院(北京 100069)
先天性糖基化障碍(congenital disorders of glycosylation,CDG)是一种罕见的常染色体隐性遗传代谢病(OMIM 608540),其由于遗传缺陷导致糖基化生物合成途径异常,可发生于蛋白质N-糖基化和O-糖基化、脂质糖基化途径等[1]。ALG 1基因变异所致的CDG(ALG 1-CDG)主要是由于ALG 1基因变异导致β-1,4甘露糖转移酶缺陷,不能完成蛋白N-糖基化途径中聚糖的组装和加工[2],可累及神经、血液、骨骼、胃肠道及眼睛等全身多个系统。目前国内仅报道2 例ALG 1-CDG 患儿[3-4],且无关于ALG 1-CDG 表现为婴儿痉挛症的报道。本文报告2 例临床表型为婴儿痉挛症的CDG 患儿的临床资料及基因变异类型,强调对此类患儿早期进行基因检测明确诊断的重要性。
1 临床资料
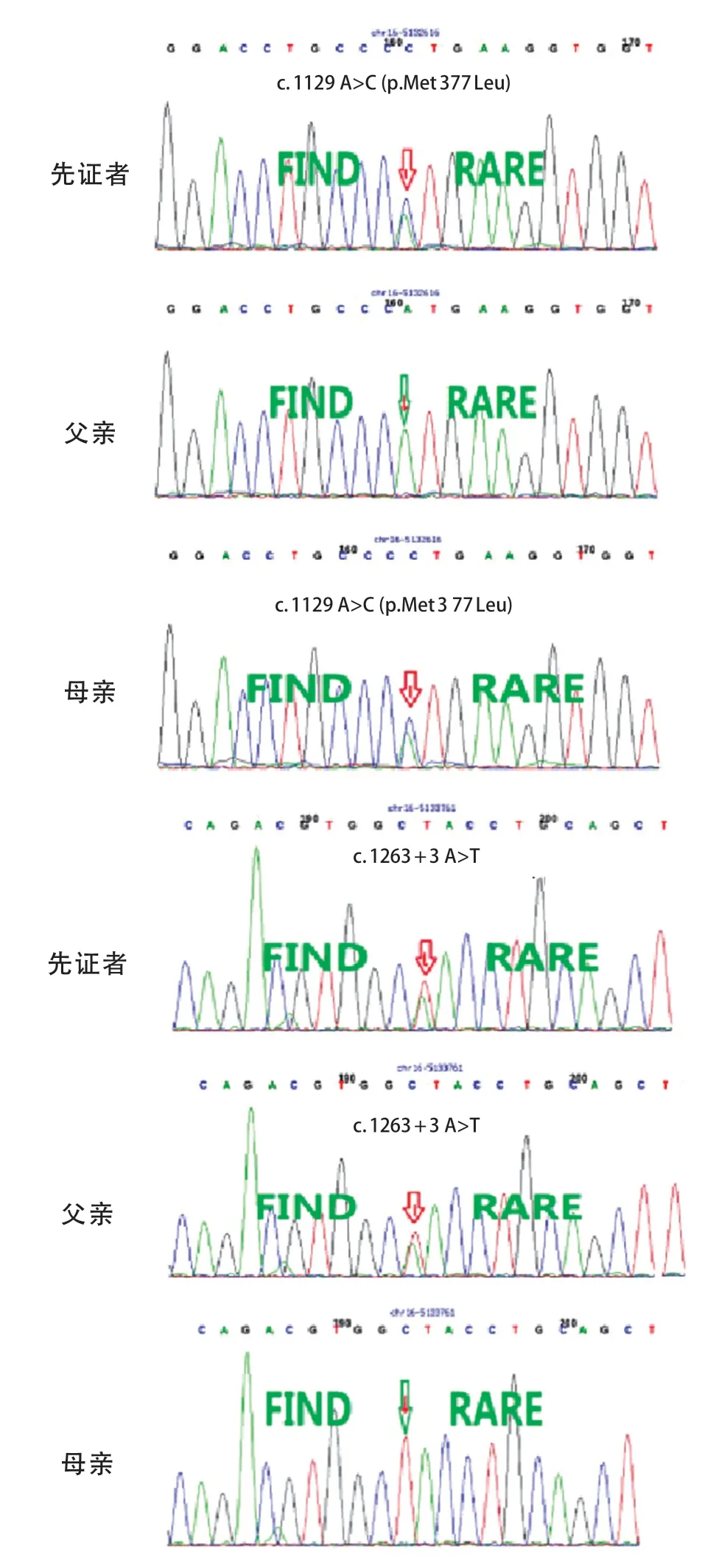
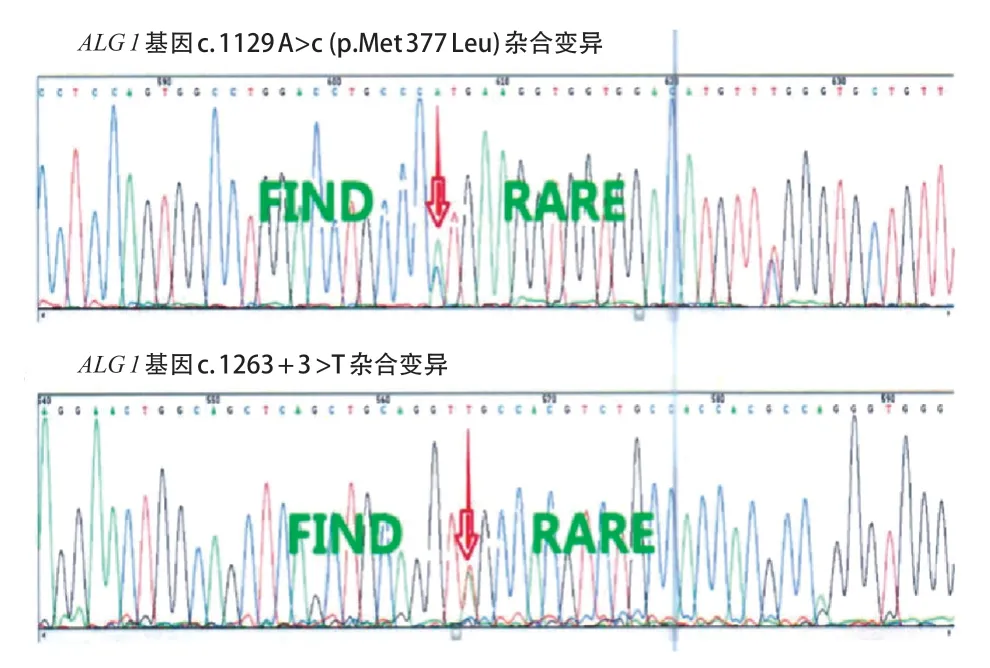
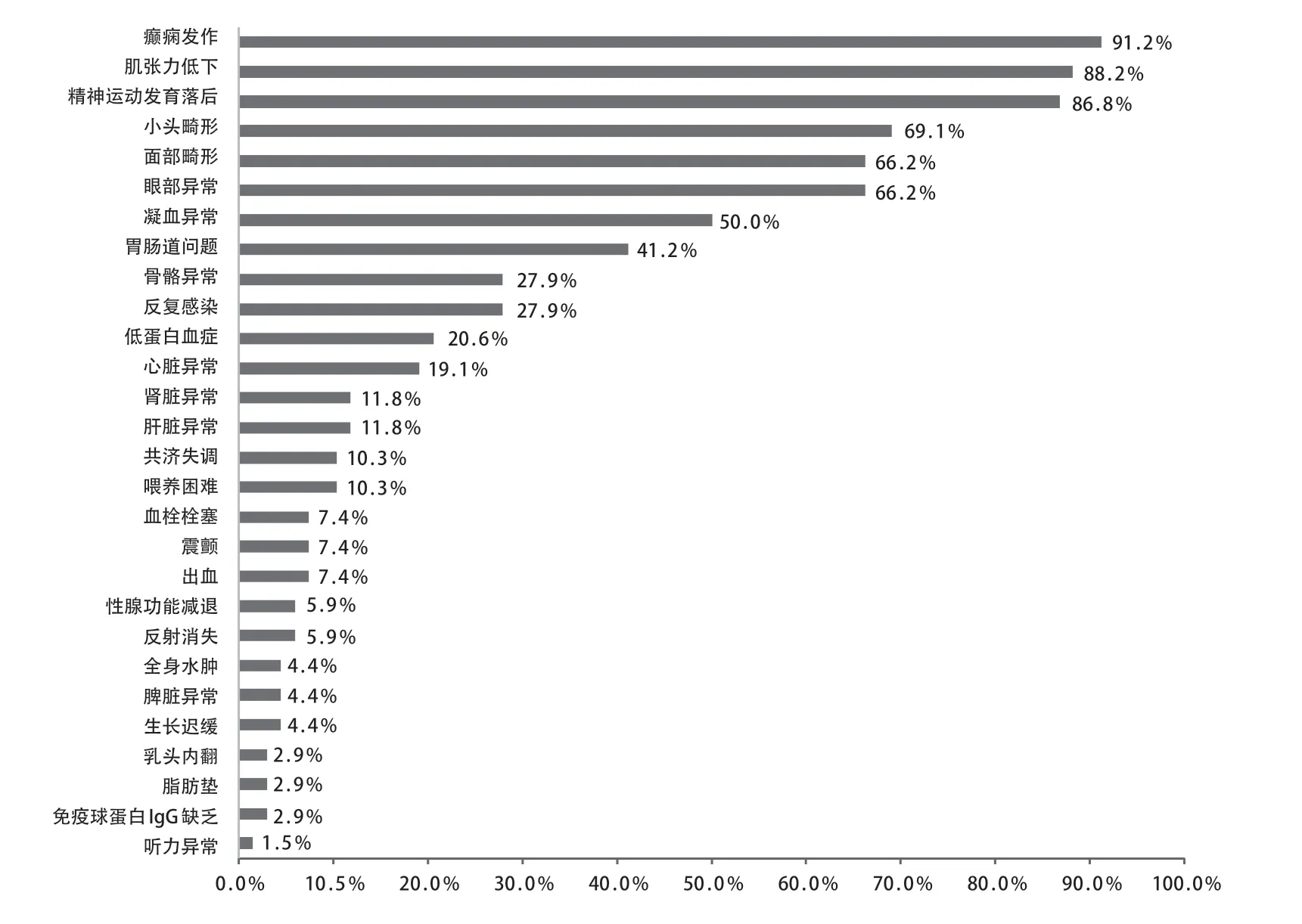
先证者,女,4 月龄,因发现追物、追声倒退1 月余、间断抽搐发作1周入院。患儿3月龄时出现追物、追声倒退,抬头不稳,抓握欠佳;4 月龄出现点头抱团样发作,成串发作,5、6 串/d,7~15 下/串。脑电图示高峰失律(图1 A),诊断为婴儿痉挛症。头颅磁共振成像(MRI)无明显异常。给予辅酶Q10、左卡尼汀、氨己烯酸口服治疗,并予促肾上腺皮质激素(adrenocorticotrophic hormone,ACTH)静脉输注,在ACTH 治疗结束后患儿发作较前减少,但脑电图高峰失律未消失。2 岁7 月龄时患儿仍存在点头抱团样发作,1、2串/d,10余下/串,且仍有明显的精神运动发育落后,不能追物追声,大运动及精细动作均较差,抬头不稳,不会爬,不会坐,不会站立,不能主动抓握,偶尔发出“ma”音。患儿系G 2 P 2,足月顺产,出生体质量3.43 kg,有新生儿ABO 溶血史(父为B型血,母为O 型血),否认围生期脑损伤史,Apgar 评分为10 分。母亲有妊娠期糖尿病史。父母非近亲结婚,有一哥哥(例2)。患儿4 月龄时体格检查:身长63 cm,体质量7.1 kg,头围不详;神志清醒,营养良好,表情呆滞,眼球运动不自主,不追物,低耳位;全身皮肤黏膜无黄染,左侧乳头内陷,全身淋巴结无异常,心肺腹无异常;脊柱无异常;四肢肌张力低,双侧肱二、三头肌腱反射正常,双侧膝、跟腱反射正常,双侧巴氏征及脑膜刺激征阴性。患儿2 岁7 月龄时头围45.5 cm( 先证者哥哥,曾于1 岁10 月龄时因间断抽搐1年6 月余入院。患儿生后即有追物、追声差,3 月龄无明显诱因出现抽搐,表现为双眼凝视、头后仰、双手握拳、四肢僵硬,持续数秒,每天10 余次。4 月龄时发作形式转为点头抱团样、双眼上翻、面部青紫,50 次/d。脑电图示高峰失律(图1 B),诊断婴儿痉挛症。视觉诱发电位(VEP)示双眼视神经传导阻滞。11月龄时曾出现血小板减少性紫癜,凝血异常。给予托吡酯、丙戊酸、左乙拉西坦、氯硝西泮及左卡尼丁口服治疗,效果不佳;进行2 次ACTH 联合硫酸镁治疗,发作减少>50%,复查EEG 仍有高峰失律。7 岁 2 月龄时患儿偶有睡醒后愣神,双手握拳。明显的精神运动发育落后,可追物、追听,但交流反应差;大运动及精细动作均较差,坐不稳,不会爬,不会站,不会主动抓东西,偶会叫发“mama”音。头颅MRI示髓鞘化延迟,T1W1 神经垂体高信号不明显,双额颞脑沟及蛛网膜下腔增宽,枕大池扩大。患儿系G1P1,足月顺产,出生体质量3.5 kg,出生时蛛网膜下腔少量出血,无其他围生期脑损伤史,Apgar 评分不详。母孕期无异常病史。1岁10月龄时体格检查身长90 cm,体质量12.3 kg,头围不详;神志清醒,营养良好,表情自然,眼球活动正常;全身皮肤黏膜无异常;心肺腹无异常;四肢肌张力偏低,双侧肱二、三头肌腱反射正常,双侧膝、跟腱反射正常,双侧巴氏征及脑膜刺激征阴性。7 岁2 月龄时体检发现患儿脊柱后凸,头围48 cm( 图1 2 例患儿脑电图表现 为明确病因,先证者4 月龄时行遗传学检查。经医学伦理审核通过,并获得法定监护人知情同意后,采集先证者及其父母外周静脉血3 mL,EDTA抗凝。从全血中提取基因组DNA,采用家系全外显子基因检测技术进行测序,使用Roche Nimblegen SeqCap EZ v5.1试剂盒(Roche,瑞士)捕获全外显子区域,采用illumina Xten(illumina 公司,美国)进行高通量测序,若检测发现可疑位点突变,采用Sanger 测序技术对父母进行验证。使用Mutation taster、PolyPhen-2及TraP Score等生物信息学预测软件预测变异位点的致病性及对蛋白功能的可能影响。结果提示先证者存在ALG 1基因复合杂合变异,即c.1129 A>C(p.Met377Leu)和c.1263+3A>T。Sanger 测序验证提示2 个变异位点分别遗传自父母(图2)。先证者哥哥经Sanger 验证与先证者存在相同的ALG 1 变异位点(图3)。变异位点c.1129 A>C(p.Met 377 Leu)为错义变异,c.1263+3 A>T 为剪切变异。两个变异位点为ESP 数据库(http:// evs.gs.washington.edu/EVS/)、千人数据库(http:// www.internationalgenome.org)、ExAC 数据库(http:// exac.broadinstitute.org)正常对照人群中未发现的变异,符合中等强度致病证据(PM 2)。两个变异位点分别来自父母,符合中等强度致病证据(PM 3)。错义变异p.Met 377 Leu 之前未曾报道,但在同一位点,导致另外一种氨基酸的变异已经确认是致病的,即Met377Val已被ClinVar数据库(http://www.ncbi.nlm.nih.gov/clinvar)收录,且报道是致病的,符合中等强度致病证据(PM 5)。位点c.1129 A>C(p.Met377Leu)经Mutation Taster及PolyPhen2预测具有可能致病性,位点c.1263+3A>T经Spidex1预测其可能影响剪切,dbscSNV 预测为有害,TraP Score得分为0.852,提示致病性可能,均符合支持致病证据(PP 3)。先证者以癫痫、精神运动发育落后、肌张力低下及小头畸形等神经系统表现为主,符合ALG 1 相关CDG 特征,且先证者哥哥存在相似的临床表现,符合支持致病证据(PP 4)。根据ACMG 标准,位点c.1129 A>C(p.Met377Leu)评级为PM2+PM3+PM5+PP3+PP4,判定为可能致病性变异。位点c.1263+3A>T评级为PM2+PM3 +PP3+PP4,判定为可能致病性变异。 图2 先证者及父母全外显子基因测序图 图3 先证者哥哥Sanger 测序结果 以“ALG 1-CDG” “CDG-Ik” “ALG 1-先天性糖基化障碍” “先天性糖基化障碍Ik 型”为检索词检索PubMed(https://pubmed.ncbi.nlm.nih.gov)、人类基因组突变数据库(HGMD,http:// www.hgmd.cf.ac.uk)、在线人类孟德尔遗传数据库(OMIM,http://omim.org)、中国知网(CNKI)和万方数据库,检索时间为建库至2020年11月30日,收集信息完整患儿的临床和遗传学资料进行分析总结,包括临床表型、基因变异类型、影像学检查及预后等。 共检索到14 篇报道ALG 1-CDG 患儿的英文文献[2,5-17],2 篇中文文献[3-4]。16 篇文献中报道70 例ALG 1-CDG 患儿,男31 例、女36 例,3 例未注明性别。其中68 例患儿有详细的临床表型信息(图4)。62例有头颅MRI信息的患儿,41例(66.1%)存在异常,其中脑或小脑萎缩22例(35.5%),皮质萎缩4例(6.5%)。在已报道文献中尚无关于婴儿痉挛症表型的报道。 图4 68 例ALG1-CDG 患儿表型谱及其比例 ALG 1-CDG 患儿预后不同,可从出生后几周内死亡到存活至成年[2,5,18]。在已报道的68 例患儿中,1 例在胎儿期即发现面部畸形,提前终止妊娠;29 例(42.6%)早期死亡,其中20例于婴儿期死亡,8例婴儿期后死亡,1例死亡时间不详。另外,p.Ser258Leu纯合变异患儿通常在出生后最初几个月死亡[2]。在Ng 等[2]报道的17 例死亡患儿中有6 例c.773 C>T(p.Ser 258 Leu)纯合变异者均在6 月龄前死亡,另外11 例早期死亡患儿中有3 例为c.773 C>T (p.Ser 258 Leu)纯合变异,均在10 月龄内死亡。 CDG 是由酶缺陷导致的聚糖合成及其与蛋白质和脂类结合的途径异常,其中蛋白质糖基化有两种主要类型,即N-糖基化和O-糖基化[19]。根据酶缺陷发生环节不同,蛋白N-糖基化障碍可分为两种类型,CDG-Ⅰ是由脂联寡糖合成缺陷及寡糖转移到蛋白质的缺陷引起,主要发生在细胞质和内质网中;CDG-Ⅱ是由蛋白质结合的聚糖的加工过程缺陷引起,主要发生在内质网和高尔基体中[2;5]。目前最常见的 N-糖基化障碍是PMM 2-CDG(CDGⅠa)[5]。CDGIk 是CDG-Ⅰ的一种亚型,是ALG 1基因变异相关的CDG,即ALG 1-CDG[2]。ALG 1基因位于16 p 13,编码β-1,4 甘露糖转移酶(OMIM 608540)。ALG 1基因变异使得甘露糖转移酶活性下降,不能完成聚糖组装或加工,进而可引起CDG,即ALG 1-CDG[2]。 ALG 1-CDG 可累及全身多系统,表现为全身水肿,凝血异常,面部畸形(低耳位、小下颌、薄嘴唇、大脑门、鼻梁扁平等),乳头内翻,脂肪垫,神经系统疾病(癫痫、精神运动发育落后、肌张力低下等),骨骼异常(脊柱侧凸、脊柱后凸、关节挛缩、髋关节发育不良),视力障碍(斜视、眼球震颤、视网膜病变及视力丧失),听力异常(耳聋),胃肠道问题(慢性腹泻、蛋白丢失性肠病、便秘),心脏损害,肝脾肿大,肾脏疾病,性腺功能低下,低蛋白血症等[2,5-9,20-21]。通过等电聚焦、毛细管区带电泳或高效液相色谱法分离血清转铁蛋白亚型,可以筛选n-链糖基化缺陷,CDG的明确诊断需要酶和基因检测[5]。 ALG 1-CDG 患儿常以严重的神经系统表现为主,如癫痫、发育迟缓、智力障碍、肌张力低下、小头畸形、共济失调等,头颅MRI 可表现为小脑发育不全、脑或小脑萎缩等[2,6,20-21]。本研究同一家系的2例患儿也是以神经系统表现为主,均有痉挛性发作,脑电图呈高峰失律,可明确诊断为婴儿痉挛症,均于出生后即存在精神运动发育落后,且存在小头畸形及肌张力低下。目前国内外尚无ALG 1-CDG 相关的明确诊断为婴儿痉挛症的报道,Pereira等[10]报道了2 例ALG 1-CDG 患儿,均表现为痉挛性发作,但其脑电图不存在高峰失律。Barba等[11]报道了1例ALG1-CDG 患儿最初表现为局灶性强直发作,后期转为痉挛性发作,但其脑电图也不存在高峰失律。对于不明原因的婴儿痉挛症患儿,当其同时存在ALG 1-CDG相关的其他神经系统表现时,如明显的精神运动发育落后、肌张力低下及小头畸形等,需考虑ALG 1变异的可能。ALG 1-CDG 是一个全身多系统疾病[2],本研究中的患儿还存在凝血异常、乳头内陷、心肌损害及身材矮小等异常。故对于婴儿痉挛症患儿,若同时存在其他多系统的表现,更应警惕ALG 1-CDG 的可能。 本研究的2 例兄妹,围生期均无明确的脑损伤病史,且头颅MRI 无明显的可解释婴儿痉挛症病因的特异性表现。妹妹与哥哥的临床表现极其相似,均存在精神运动发育落后及IS,父母非近亲婚配且无类似家族史,考虑存在严重复合杂合的隐性遗传病、母系遗传性疾病可能,对先证者及其父母先行全外显子基因检测,结果提示ALG 1基因变异,且具有致病性,随后证实先证者哥哥存在相同基因位点变异,遗传模式符合孟德尔遗传定律。神经系统疾病具有高度的遗传基础,近年来随着遗传检测技术的快速发展,外显子检测、染色体微阵列、甲基化检测、全基因组检测费用显著降低,实现了临床遗传学检测的普及化。对于存在神经系统异常的患儿家庭,在准备再次生育前,均应进行充分的遗传学检查,尽可能明确具体病因后再考虑生育。对当时未能明确病因者,在二次生育前应对先证者遗传数据根据最新遗传数据库及生物信息分析方法进行数据重分析,且应对再次孕育的胎儿进行全外显子等遗传学检测及其与先证者的遗传关联分析,进一步减少漏诊可能。充分的产前遗传咨询及产前诊断对避免重复的悲剧发生在同一个家庭内具有重要的意义。 对既往文献的总结显示,ALG 1-CDG 患儿以癫痫、精神运动发育落后、肌张力低下及小头畸形等神经系统表现最常见。本研究首次提出婴儿痉挛症可作为ALG 1-CDG 相关的癫痫患儿的表型。ALG 1-CDG 患儿预后较差,总结已有报道得出其存在约42.6%的病死率,其中以c.773C>T(p.Ser258Leu)变异位点致死率最高。 综上,ALG 1-CDG 以严重的神经系统表现为主,婴儿痉挛症可作为临床表型之一。对于婴儿痉挛症患儿,当其同时存在明显的精神运动发育落后、小头畸形及肌张力低下等神经系统表现时需要考虑ALG 1-CDG 的可能,尤其存在面部畸形或累及眼部、胃肠道、骨骼等全身多个系统时,更应警惕ALG 1 变异的可能,尽早进行基因检测。ALG 1-CDG 预后较差,致死率高,早期明确诊断有助于做好下一步的遗传咨询,优生优育。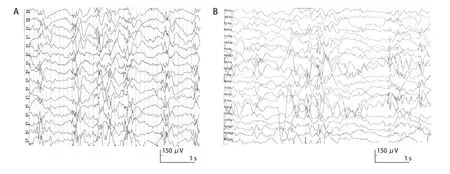
2 讨论
 猜你喜欢
临床输血与检验(2022年3期)2022-06-22实用肝脏病杂志(2022年2期)2022-03-21中国康复(2021年6期)2021-11-30复旦学报(医学版)(2021年5期)2021-10-13三农资讯半月报(2020年8期)2020-05-13诊断学(理论与实践)(2020年1期)2020-04-28家庭百事通·健康一点通(2017年8期)2017-08-18中国实用医药(2016年9期)2016-05-17医学研究杂志(2015年7期)2015-06-22医学研究杂志(2015年12期)2015-06-10
猜你喜欢
临床输血与检验(2022年3期)2022-06-22实用肝脏病杂志(2022年2期)2022-03-21中国康复(2021年6期)2021-11-30复旦学报(医学版)(2021年5期)2021-10-13三农资讯半月报(2020年8期)2020-05-13诊断学(理论与实践)(2020年1期)2020-04-28家庭百事通·健康一点通(2017年8期)2017-08-18中国实用医药(2016年9期)2016-05-17医学研究杂志(2015年7期)2015-06-22医学研究杂志(2015年12期)2015-06-10
