
S-EPA及其羧酸酯前药在犬体内的药动学
2021-07-16 09:08张雨沐孙钰菲刘兴华王文艳
烟台大学学报(自然科学与工程版) 2021年3期
李 鑫,张雨沐,孙钰菲,刘兴华,王文艳
(烟台大学药学院,新型制剂与生物技术药物研究山东省高校协同创新中心、分子药理和药物评价教育部重点实验室,山东 烟台 264005)
S-EPA为硫取代epacadostat (EPA)[1]的类似物,是一种有效的小分子吲哚胺2,3-双加氧酶(Indoleamine 2,3-Dioxygenase, IDO)抑制剂。IDO可催化色氨酸(teyptophan,Trp)转化为犬尿氨酸(kynurnine,Kyn),其过度表达与肿瘤免疫逃逸密切相关[2-6]。实验室前期研究结果表明S-EPA与EPA(epacadostat)对荷瘤小鼠的IDO活性抑制率基本一致[7]。前药设计可增加口服或者局部给药的吸收度、提高生物利用度等,已成为一种有效策略而广为接受[8-11]。李红等设计了夫西地酸(Fusidic Acid,FA)羧酸酯前药 F1 和 F2,在大鼠给药 FA、F1、F2 后,其中 F1 的吸收程度更高,具有更高的生物利用度[12]。加巴喷丁酯是加巴喷丁的消旋体前药,在大鼠和猴子中口服后,被有效吸收并迅速转化为加巴喷丁[13],在临床使用中,加巴喷丁酯可以通过提高疗效、减少患者间差异和减少给药频率来改善神经性疼痛、癫痫病和许多其他疾病的治疗[14]。对于吸收较差、生物利用度低的药物,可尝试采用设计为酯类前药的手段改善化合物的缺陷。在犬单次灌胃给予S-EPA和EPA后,S-EPA在体内的暴露量低于EPA,约为其70%。因此本研究设计了一种S-EPA的羧酸酯前药PCC1,结构如图1所示,旨在改善S-EPA的吸收度,提高其生物利用度。

图1 PCC1化学结构式
1 实验材料
1.1 主要仪器
AB 4500-Qtrap三重四级杆串联质谱仪(美国AppliedBiosystems公司),配备有ESI 离子源;ExionLCTMAD系列高效液相色谱仪(美国AppliedBiosystems公司);Q Exactive Orbitrap高分辨质谱(美国ThermoFisher公司);核磁共振(Nuclear Magnetic Resonance, NMR)谱采用Avance Neo 600 超导核磁共振仪(德国Bruker公司)测定,1H-NMR、13C-NMR 的测定频率分别为 600 MHz、150 MHz。
1.2 药品与试剂
EPA (上海升德医药科技有限公司,纯度>99%);S-EPA(烟台大学药物化学实验室自制,纯度>98%);PCC1 (实验室自制,纯度>95%);甲醇、乙腈(色谱纯,德国Merck);Solutol、乙酸铵(美国sigma公司);甲基叔丁基醚、二氯甲烷(分析纯,国药集团化学试剂有限公司);实验用水为超纯水。
1.3 实验动物
雄性比格犬6只,体重为10 kg左右,由山东绿叶制药有限公司实验动物中心提供,许可证号 SCXK(鲁) 20170006,所有动物都严格按照美国国立卫生研究院实验动物护理和使用指南自由获取饲料和水。动物相关的所有实验均经烟台大学实验动物伦理委员会审核并批准。
2 实验方法
2.1 色谱质谱条件
色谱条件:色谱柱为shim-pack,XR-ODS C18柱(3.0 mm×50 mm, 2.2 μm)。
液相条件:流动相为85% 乙腈-15% 水,含0.4 mmol·L-1醋酸铵;进样体积5 μL;流速为0.3 mL·min-1;柱温为30 ℃。
质谱条件:电喷雾离子化电离源(ESI),负离子检测模式:离子化电压-4500 V,毛细管温度450 ℃,气帘气30 psi,喷雾气55 psi。两种分析物及内标的LC-MS/MS参数见表1。

表1 两种分析物及内标的LC-MS/MS 参数
2.2 标准溶液的制备
分别精密称定PCC1、S-EPA各2份,用适量甲醇溶解稀释配制成浓度为1 mmol·L-1的储备液。分别取储备液适量,用甲醇连续稀释以制备所需浓度的标准工作液和质控工作液,内标用甲醇稀释至5 μmol·L-1待用。
2.3 全血样品处理方法
精密量取冰水浴下融化的犬全血样品 45 μL,加入5 μL内标(EPA, 5 μmol·L-1),精密加入500 μL 乙腈,涡旋1 min后,超声10 min,以13 000 r/min离心10 min,取上清液加入提取液(叔丁基甲醚∶二氯甲烷=3∶2,V/V),涡旋3 min后,4000 r/min离心10 min,取上清液于37 ℃水浴氮吹,吹干后100 μL流动相复溶,进样5 μL分析。
2.4 方法学验证
根据《中国药典》(2015 版)[15]对于生物样品定量分析方法学验证的指导原则,针对S-EPA血药浓度测定的 LC-MS/MS 方法进行系统的方法学考察,内容包括专属性、线性范围和定量限、提取回收率、精密度与准确度、基质效应以及不同条件下的样品稳定性。
2.5 犬药代动力学实验
比格犬禁食不禁水 12 h 后随机分为 2 组,即PCC1 和S-EPA 组,每组3 只,分别灌胃给予 PCC1和S-EPA的 20% Solutol 溶液,给药剂量为 5 mg/kg,给药体积为1 mL/kg,分别于给药前及给药后 15 min、30 min、1 h、2 h、3 h、4 h、6 h、8 h、12 h、24 h静脉采血约1 mL置于肝素化EP管中,-20 ℃冻存待测。
2.6 统计学方法

3 结果与讨论
3.1 PCC1结构确认
经高分辨质谱扫描其m/z[M+H]+=496.969 82,其理论值为495.963 45,偏差在2.94。PCC1的1H-NMR和13C-NMR结果见表2,结果表明该化合物为所合成的目标化合物PCC1。

表2 PCC1用氘代二甲基亚砜溶解后的 NMR结果
3.2 方法学验证
3.2.1 专属性 通过分析来自6个不同个体的空白全血考察方法的专属性。结果表明,在测定条件下, 全血中内源性物质不干扰分析物的测定,代表性色谱图如图2所示。

图2 LC-MS/MS测定犬全血样品中S-EPA药物浓度的典型色谱图
3.2.2 线性范围、定量下限与残留 取室温下融化的空白犬全血45 μL,加入5 μL内标(EPA, 5 μmol·L-1)分别精密加入不同浓度S-EPA对照品工作液 5 μL(使全血中的S-EPA的浓度分别为10、20、40、80、200、400、800、2000 nmol·L-1,再精密加入500 μL乙腈,其余操作步骤同样品处理过程,同时制备一个空白样品和一个零浓度样品(含内标)。以待测药物浓度为横坐标,待测物与内标物的峰面积比值为纵坐标,用加权(W=1/X2)最小二乘法进行回归运算,三天的直线回归方程分别为
Y=0.003 44x+0.007 46,
Y=0.002 65x+0.001 21,
Y=0.003 1x-0.001 04,
相关系数R分别为0.994 3、0.996 9、0.996 0。结果S-EPA全血药物浓度在10~2000 nmol·L-1范围内线性关系良好,R>0.994 3。定量下限为10 nmol·L-1。在最高浓度样品(含内标)进样后空白样品中的分析物和内标保留时间处均无明显色谱峰(即待测物峰面积小于定量下限的20%,内标峰面积小于5%),表明系统无分析物及内标残留。
3.2.3 提取回收率与基质效应 按样品处理方法制备低、高浓度的质控样品,并且制备内标和低/高浓度标准工作液加入到经预处理的6 个不同来源的空白犬全血中配制成相当于低、高质控浓度的样品,以及制备相应浓度的标准工作溶液,分别比较提取样品中每种分析物和内标的峰面积比与含基质工作液中峰面积比值,计算提取回收率,比较含基质工作液与不含基质工作溶液中的峰面积比考察基质效应。每个浓度平行6份,化合物回收率、基质效应结果见表3。
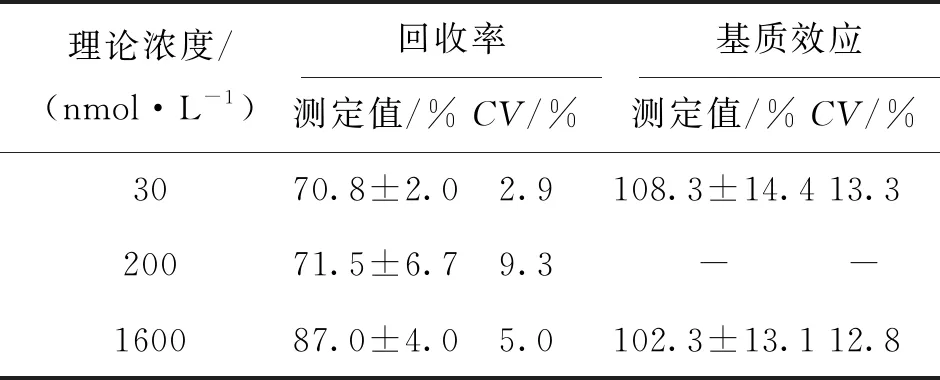
表3 犬全血中S-EPA的提取回收率及基质效应(n=6)
3.2.4 精密度与准确度 配制定量下限、低、中、高(10、30、200、1600 nmol·L-1) 4个浓度的 S-EPA 标准全血样品各6 份,按“2.3”项下方法处理、测定,得日内变异;每日配制定量下限、低、中、高4个浓度的S-EPA标准全血样品各6份,连续3 d测定,得日间变异,并计算准确度。如表4所示,S-EPA定量下限的批内、批间精密度(用CV表示)均小于7.6%,准确度(用RE表示)均小于6.6%。表明该方法的精密度和准确度符合生物样品测定要求。

表4 LC-MS/MS法测定犬全血中S-EPA的精密度和准确度
3.2.5 稳定性 取空白犬全血血浆45 μL,加入低、高浓度的标准溶液配制低、高浓度质控,考察犬全血样品自动进样器放置24 h、3次冻融循环的稳定性,-20 ℃下放置14 d的稳定性。每种条件下每浓度3个平行样本。稳定性考察实验结果如表5所示,S-EPA在各条件下稳定性良好,样品测定和保存条件下的稳定性均符合生物样品测定要求。

表5 不同条件下S-EPA在犬全血中的稳定性(n=3)
3.3 犬体内药动学评价
健康雄性比格犬在 5 mg/kg 剂量下单次灌胃给予 S-EPA和PCC1 后,PCC1迅速转化为 S-EPA,全血中未检测到该前药,其S-EPA 的平均血药浓度-时间曲线见图 3。根据血药浓度经时变化曲线数据,采用 Winnonlin 8.1软件非房室模型法计算各给药组 S-EPA的药动学参数,结果见表6。比格犬灌胃给药 S-EPA和PCC1后,PCC1 组Cmax提高了1.54倍,改善了S-EPA的吸收,与S-EPA组比较,生物利用度是其135%。

表6 犬灌胃给予S-EPA及PCC1后平均全血S-EPA药代动力学参数(n=3)

图3 犬灌胃给予 S-EPA和PCC1后S-EPA在犬全血中的平均药-时曲线(n=3)
4 结 论
本文建立了灵敏、快速、准确的 LC-MS/MS 方法用于犬全血中 S-EPA药物浓度测定,并进行了系统的方法学验证,符合S-EPA血药浓度测定的生物分析要求,并且方法成功应用于S-EPA的羧酸酯前药与S-EPA的犬体内药代动力学特征评价。结果显示,羧酸酯前药 PCC1 较原型药物 S-EPA 在吸收方面有明显改善,提高了其在犬体内的生物利用度。
猜你喜欢
人人健康(2022年18期)2022-10-10
中外医学研究(2022年22期)2022-09-09
人人健康(2022年13期)2022-07-25
检验医学与临床(2022年1期)2022-01-26
口腔护理用品工业(2021年4期)2021-11-02
临床内科杂志(2021年9期)2021-09-17
中国科技纵横(2019年23期)2019-02-14
中国当代医药(2018年12期)2018-06-16
人人健康(2017年13期)2017-07-26
海峡科技与产业(2017年1期)2017-03-04
