
以肝硬化合并急性腹痛为表现的红细胞生成性原卟啉病1例
2021-07-13 08:23:40刘晓燕苏海滨
传染病信息 2021年3期
李 会,李 晨,刘晓燕,苏海滨
1 病例报告
1.1 病史 患者,男,40岁,已婚,山西人,煤矿办公室工作人员,因“间断乏力、身目黄染2年余,加重伴腹痛6 d”于2018年7月4日入住中国人民解放军总医院第五医学中心。患者2015年11月出现乏力,身目黄染,尿黄如豆油色,后逐渐加重,2016年2月于我中心就诊,诊断为“肝硬化失代偿期合并腹水(肝硬化原因不明)”,予对症保肝等治疗后症状好转出院。2018年3月患者再次出现上述症状,自行口服“中药(具体不详)”治疗2月余,无效。6月27日上述症状加重,并伴有腹痛,疼痛部位不固定,有时放射至右侧胸部及后腰部,持续性钝痛、针扎样痛、绞痛、烧灼样疼痛交替,无明显缓解及加重因素。6月30日再次就诊于我中心,查肝功能异常,遂以“肝硬化失代偿期”收入病房。患者既往约10岁时出现“皮肤病(自诉为日光过敏性皮炎,表现为暴露部位的烧灼样感、红肿、水泡、结痂)”,间断口服“中药、西药”等治疗,效果不佳,仍反复发作,患者哥哥及其女儿也有类似表现(较患者表现轻微),父母非近亲结婚,无其他基础疾病及遗传病诊断史,无烟酒史及吸毒史,育有1女1子,1子为“白化病”患儿,女儿、爱人及父母均身体健康。
1.2 入院查体 生命体征平稳,营养中等,神志清楚,精神差,面色晦暗,皮肤、巩膜轻度黄染,口周及双上肢可见片状结痂性皮肤(光过敏皮肤损伤),肝掌阳性,蜘蛛痣阴性。全身浅表淋巴结未触及肿大。心肺未见异常。腹部平软,未见腹壁静脉曲张,右下腹压痛明显,轻微反跳痛,肝右肋下2 cm可触及,缘钝,质中,无触痛,莫菲氏征阴性,脾左肋下未触及。移动性浊音可疑阳性,肠鸣音正常。双下肢无水肿。神经系统查体未见异常。
1.3 实验室检查及其他辅助检查 腹水常规未提示感染(腹水常规:颜色黄、透明度清、李瓦它试验阴性、细胞总数1146×106/L、WBC 146×106/L、中性粒细胞百分比23%、间皮细胞百分比8%、淋巴细胞百分比69%),CRP 33.5 mg/L,降钙素原1.08 ng/ml。肝功 能:ALB 40 g/L、T/DBIL 201.3/158.8 µmol/L、ALT 216 U/L、AST 458 U/L、ALP 233 U/L、谷氨酰转肽酶(γ-glutamyl transpeptadase, GGT)584 U/L,γ- 球蛋白 19.5%,IgG 18.39 g/L,铜兰蛋白0.43 g/L,铁46.7 µmol/L。凝血功能:凝血酶原活动度(prothrombin time activity, PTA)58.9%、国际标准化比值(international normalized ratio, INR)1.22。血常规:WBC 5.12×109/L、HGB 127.00 g/L、中性粒细胞百分比75.6%、PLT 25.00×109/L。甲状腺功能正常。甲、乙、丙、戊型肝炎病毒标志物阴性,单纯疱疹病毒、EB病毒、巨细胞病毒相关血清学抗体阴性。抗核抗体及肝病自身抗体均阴性。腹部增强CT:①肝硬化、脾大、副脾、腹水、食管及胃底静脉曲张、脾静脉曲张。②胆囊炎。③肝囊肿,右肾囊肿。④肝动脉变异。肺部CT:左肺上叶陈旧性病变。腹部立位平片:不完全肠梗阻,可见气液平面。肝脏血管超声未见异常。泌尿系统超声未见异常。盆腔CT:盆腔积液、腹腔积液、盆腔钙化灶。
1.4 诊治经过 入院后结合患者病史、查体及辅助检查,排除病毒性、自身免疫性、酒精性等常见肝损伤病因,并查阅了相关遗传性代谢性肝损伤的文献[1-2],排除肝脏血管病变、肝豆状核变性、铁代谢异常血色病及抗胰蛋白酶缺乏症等先天性遗传代谢性肝损伤疾病。肝硬化原因有待进一步明确,腹痛原因为肠梗阻及腹膜炎(临床诊断),并考虑肠梗阻为腹痛的主要原因。治疗上给予禁食水、胃肠减压、石蜡油灌肠、抗感染、保肝、降酶、退黄、抗淤胆等内科综合治疗,效果不佳,患者腹痛无明显缓解,疼痛范围逐渐扩展至腰背部、膀胱区、前胸、后背、上臂及大腿,疼痛性质同前,肝功能及凝血功能进行性恶化,进展为肝衰竭。7月18日复查肝功能:ALB 30 g/L、T/DBIL 437.6/324.3 µmol/L、ALT 251 U/L、AST 480 U/L、ALP 216 U/L、GGT 271 U/L。凝血功能:PTA 38.8%、INR 1.60。血常规:WBC 6.12 ×109/L、HGB 90.00 g/L、中性粒细胞百分比82.8%、PLT 36.00×109/L。再次重新分析病情,结合患者既往有日光性皮炎、哥哥及其女儿有类似皮肤病,此次发病主要以肝功能异常、腹痛、肠梗阻为主要临床表现,抗生素及胃肠减压等治疗无效,查体见口周破溃,呈放射状萎缩性纹理(图1),双手部皮肤粗糙(图2),考虑卟啉病。
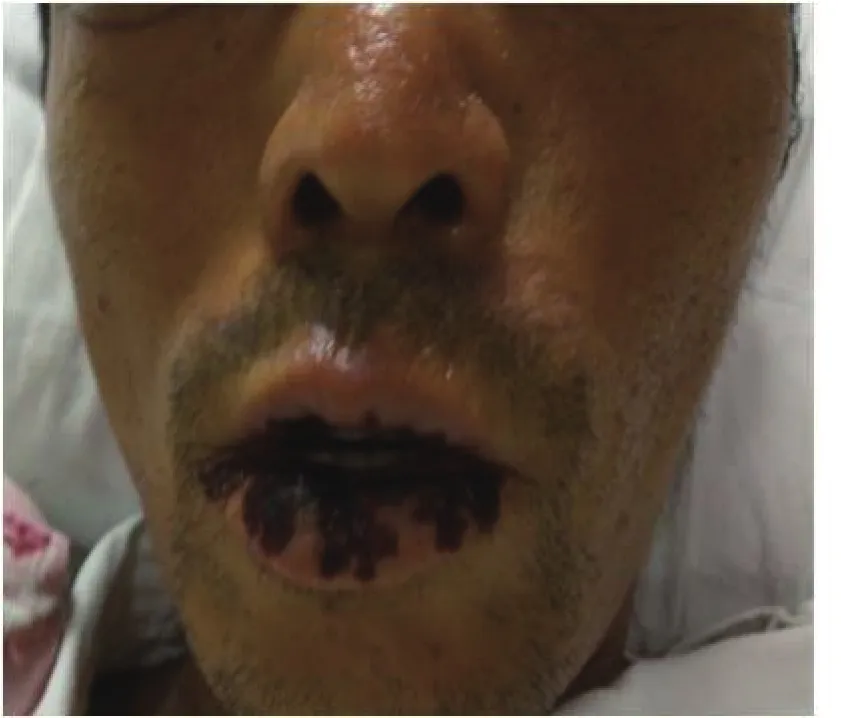
图1 患者口唇部皮肤表现口周破溃,呈放射状萎缩性纹理Figure 1 Skin manifestations on patient’s lip and mouth

图2 患者手部皮肤表现桡侧及虎口处皮肤增厚,手背皮肤粗糙Figure 2 Skin manifestations on patient’s hand
1.5 确诊 2018年7月20日北京协和医院尿卟啉检测结果为阴性,红细胞内游离原卟啉的检测结果:细胞内锌卟啉为57.5 µg/gHb(参考值为0~4.7 µg/gHb)。患者尿液放置曝光以及在WOOD灯下无颜色变化。血液基因学检测:常染色体隐性红细胞生成性原卟啉病(erythropoietic protoporphria,EPP)相关基因亚铁螯合酶外显子及内含子区域存在3处杂合突变,即 c.892C>T(胞嘧啶>胸腺嘧啶)(图3A),c.68-23C>T(胞嘧啶>胸腺嘧啶)(图3B),c.315-48T>C(胸腺嘧啶>胞嘧啶)(图3C)。最终明确诊断:EPP。7月21日患者自动出院,于当天死亡。

图3 患者亚铁螯合酶基因突变点检测结果A.c.892C>T(胞嘧啶>胸腺嘧啶);B.c.68-23C>T(胞嘧啶>胸腺嘧啶);C.c.315-48T>C(胸腺嘧啶>胞嘧啶 )Figure 3 Detection results of FECH gene mutation points in patient
2 讨 论
卟啉病是在血红素生物合成途径中,因某些酶的缺乏或异常,引起未转化为血红素的卟啉及(或)卟啉前体(如δ-氨基-γ-酮戊酸和胆色素原)在体内过度分泌及蓄积,并在组织中沉积,最终由尿和粪便排出的一组遗传代谢性疾病。血红素在哺乳动物组织中通过受酶调控的8个步骤进行合成,根据血红素生成步骤中所需酶的不同,将卟啉病分为8个类型,每一类型均与血红素合成通路上的一种酶缺陷有关,并有相应的基因突变位点,当基因位点发生突变后可形成不同类型的卟啉病,表现为卟啉、卟啉前体或者两者特征性的蓄积和异常分泌。各类型卟啉病有其特异的相关酶缺乏的病理生理机制,基因学检测出相应的酶学基因突变点为诊断卟啉病的最直接证据。遗传方式可能为常染色体显性或隐性遗传。
EPP是因血红素合成最后一步中的催化酶亚铁螯合酶活性缺陷或活性低下,使体内原卟啉IX水平升高并在体内蓄积沉积于皮肤等全身组织所致的疼痛性遗传性皮肤病[3]。大部分EPP是由于亚铁螯合酶一个等位基因突变所致的具有不同外显率的常染色体显性遗传病。亚铁螯合酶基因已被定位于染色体18q21.3-22区域,含有11个外显子和10个内含子,少数患者(占4%)以常染色体隐性方式遗传,常染色体隐性遗传是发生肝衰竭的危险因素[4]。因染色体突变所致亚铁螯合酶缺乏,从而使原卟啉在红细胞、肝脏、皮肤中过度沉积而致病。原卟啉是一种由肝脏分泌的亲脂性分子,因此EPP患者有胆石症和胆道梗阻性发作的风险,并具有较高的肝衰竭风险,为EPP患者发病率和病死率较高的主要原因[5]。
本例患者基因学检测亚铁螯合酶存在3处杂合突变,突变位点c.892C>T(胞嘧啶> 胸腺嘧啶)为致病突变,导致蛋白翻译终止;突变位点c.68-23C>T(胞嘧啶>胸腺嘧啶)为可疑致病突变,影响蛋白剪切;突变位点c.315-48T>C(胸腺嘧啶>胞嘧啶)为疾病相关多态性位点,常常和明确致病突变点组合成复合杂合突变,从而导致EPP[6-7]。此3处基因突变点分别位于两条染色体上,且每条染色体上均有一处突变为致病性突变,结合患者病史特点及细胞内锌卟啉明显升高,故可以明确诊断EPP。但在诊断上,尚存在不足之处:①该病例因患方原因未进一步明确患者父母及子女的基因学检测以充分验证该诊断,同时未对该患者进行肝穿刺病理活检以明确肝脏病理;②在鉴别诊断中,未进行充分的基因检测和进一步检查以明确排除抗胰蛋白酶缺乏症和药物性肝损伤的可能;③在诊疗过程中,未能及时准确地明确肝硬化的病因诊断,丧失最佳治疗时机,加之花费较高,导致患者最终死亡。
EPP多在儿童期发病,比较罕见,也可迟至成年发病,男性多于女性,发病率波动在1/75万~1/20万[8]。EPP的临床表现主要有以下几个方面:①光敏性:为无水泡的急性疼痛性光敏反应,儿童时期或更早时期发病,从第一次照射阳光开始,持续终生,最初表现为阳光照射下暴露区皮肤刺痛、红斑、水肿及灼伤,持续数小时至数天,与阳光照射的强度和持续时间有关,反复的光敏性发作导致皮肤出现蜡质或皮革状增厚及角化过度外观,嘴唇呈线萎形缩性皱纹和伪横纹。其主要机制为原卟啉在血液循环及皮肤中沉积,吸收光能后呈激发状态,与氧结合生成活性氧,导致膜蛋白的交联、膜脂质的过氧化,从而使细胞膜和线粒体成为原卟啉诱导损伤的主要靶点,而整个反应过程中肥大细胞释放的5-羟色胺及花生四烯酸促进光反应的进行。②腹痛:主要表现为发作性的绞痛、胀痛、针扎样和/或刀割样交替疼痛,疼痛局限或放射至腰背部,伴有恶心呕吐、顽固性便秘甚至肠梗阻,应用止痛药有效。其发病机制可能为胃肠组织内5-羟色胺增加、胃肠自主神经功能紊乱、原卟啉毒性作用直接刺激胃肠道平滑肌。本例患者有明显腹痛、肠梗阻表现,并且有放射至膀胱区、前胸、后背部、上臂及大腿部烧灼样的疼痛,常易被误诊为“腹痛原因待查”,该患者在诊疗过程中也考虑肠梗阻及腹膜炎为腹痛原因,但经积极抗感染及解除肠梗阻治疗后,疼痛并未得以缓解。③肝损伤:大约5%~20%的EPP患者出现肝损伤表现[9]。国内外文献均有相关肝胆系统受累的EPP患者的报道,并且均发生了严重的肝损伤,甚至肝衰竭[10-14]。还有研究表明较高的红细胞原卟啉水平是EPP患者病情严重程度和肝损伤加重的主要决定因素[15]。其发病机制为原卟啉沉积于肝细胞、巨噬细胞、库弗细胞和胆管系统,在偏差显微镜下呈双折射性的晶状体,过多的原卟啉在肝脏分泌入胆汁过程中,沉积于肝细胞及胆管腔内并聚集成小晶状体,从而阻塞胆道并损伤肝细胞,而且原卟啉在肝脏淤积后易形成胆结石。原卟啉具有直接的肝毒性,当原卟啉生成过多超过肝脏代谢时,在肝脏的长久蓄积可诱发肝损伤和肝硬化。另外,其他类型的原卟啉病经常有合并神经精神症状,可有周围神经、自主神经、中枢神经系统功能及精神状态的损害,其发病机制目前尚未明确,可能与卟啉前体物质破坏神经组织有关,并未发现与原卟啉相关,所以临床上EPP患者的神经精神症状并不常见。
目前尚无根治EPP的有效治疗方案,以降低光敏感性、减少原卟啉产生、促进原卟啉排出为主,辅以其他对症支持治疗缓解症状。本病的基础治疗是控制光敏感,避免光损伤,口服胡萝卜素降低光敏性,欧洲联盟批准光损伤保护剂Afamelanotide用于治疗EPP患者的皮肤损伤[16-17]。口服鹅脱氧胆酸能降低体内原卟啉水平。考来烯胺可阻断原卟啉的肠肝循环,促进原卟啉从粪便中排出。熊去氧胆酸也可用于治疗EPP[18]。还有研究者采用反复输血提供血红素,血浆置换清除过多的原卟啉来治疗EPP合并急性胆汁淤积[19]。因肝移植并不能改变患者基因变异所致的酶缺陷,移植后仍可出现过多的原卟啉沉积于肝脏,造成肝损伤,因此肝移植并不作为推荐。肝和骨髓联合移植应被认为是一种适合严重肝受累的EPP病例的治疗方法[8]。骨髓或造血干细胞移植、干细胞基因治疗有待进一步深入研究[20]。
EPP是一种罕见的遗传代谢性疾病,临床表现形式有多样性、间歇性、反复性、渐进性、隐匿性特点,首诊形式多样,在皮肤科、普外科、神经科、消化科、血液科、儿科、肝病科均有病例报道。本例患者以肝损伤病因不明首诊,在诊疗初期皮肤损伤症状被忽视或被认为与此病无关;在病情进展阶段,并发了急性腹痛,以积极对症治疗腹痛(腹膜炎及肠梗阻)后,肝功能仍在持续恶化;在重新系统筛查肝损伤病因,结合皮肤损伤表现、腹痛特点及查阅相关文献后才考虑为EPP,临床诊疗过程曲折,极易造成疾病的漏诊、误诊。
综上所述,通过本例EPP的诊疗,丰富了该病的诊疗经验,充分意识到只有对卟啉病可能引起的皮肤损伤、腹痛、肝硬化甚至肝衰竭有所认识,形成将皮肤损伤、腹痛、肝损伤进行“一元论”的临床思维,才能做出正确的临床判断,选择合理的临床辅助检查手段,达到早期诊断及治疗,从而提高临床医生对卟啉病的认识和诊疗水平。
猜你喜欢
食品工业科技(2023年4期)2023-02-14 10:12:58
昆明医科大学学报(2021年8期)2021-08-13 08:59:04
吉林大学学报(理学版)(2021年3期)2021-05-26 02:24:00
中国食品学报(2019年10期)2019-11-12 11:31:36
武警医学(2018年10期)2018-11-06 07:04:34
当代化工研究(2016年6期)2016-03-20 16:21:42
百科知识(2015年13期)2015-09-10 07:22:44
质谱学报(2015年5期)2015-03-01 03:18:25
中国洗涤用品工业(2015年2期)2015-02-28 19:02:00
无机化学学报(2014年3期)2014-02-28 17:30:55
