
喹唑啉类表皮生长因子受体酪氨酸激酶抑制剂药物设计策略
2021-07-04 02:53:06赵永杰胡瑞娟郄正刚
煤炭与化工 2021年5期
赵永杰,孟 建,胡瑞娟,郄正刚
(河北常山生化药业股份有限公司,河北 石家庄050800)
0 引 言
酪氨酸激酶(TKs)是催化三磷酸腺苷(ATP)上的γ-磷酸基与蛋白质的酪氨酸残基羟基发生磷酸化的一类激酶。酪氨酸激酶分为受体酪氨酸激酶(RTKs)和非受体酪氨酸激酶(NTKs)。
临床研究发现,肿瘤的生成与TKs的异常表达密切相关,因此,通过抑制TKs,可以实现对肿瘤的预防和治疗。
目前,国内外已有多种以TKs为靶点的酪氨酸激酶抑制剂(TKIs)类药物批准上市。如表皮生长因子受体(EGFR)抑制剂、成纤维细胞生长因子受体(FGFR)抑制剂、血管内皮生长因子受体(VEGFR)抑制剂、间变性淋巴瘤激酶(ALK)抑制剂等。
EGFR家族是酪氨酸激酶中最早被发现的受体酪氨酸激酶。表皮生长因子受体酪氨酸激酶能够介导多条信号转导通路,将胞外信号传递到细胞内,对细胞增殖、分化和凋亡起着重要的调节作用,选择性抑制表皮生长因子受体介导的信号转导途径,可以达到治疗肿瘤的目的,这为肿瘤的治疗开辟了途径。
EGFR靶向药物主要有2类:一类是作用于受体细胞外区的单克隆抗体(MAb);另一类是作用于受体细胞内区的小分子酪氨酸激酶抑制剂。
20世纪90年代中期,Fry等人发现4-苯胺基喹唑啉(PD153035)为EGFR酪氨酸激酶的专一抑制剂,喹唑啉衍生物成为表皮生长抑制剂研发的重要方向。化合物PD153035被广泛用作EGFR抑制剂设计的先导化合物。小分子酪氨酸激酶抑制剂根据在体内结合能力的差异,可以将这类药物划分为可逆抑制剂和不可逆抑制剂。
目前,已上市的喹唑啉类小分子靶向酪氨酸激酶抑制药物有吉非替尼、厄洛替尼、埃克替尼、阿法替尼、达克替尼等。
1 EGFR酪氨酸激酶可逆抑制剂
芳胺基喹唑啉类EGFR酪氨酸激酶可逆抑制剂可模拟ATP的腺嘌呤环,通过与EGFR激酶域ATP结合位点上的ATP竞争,干扰EGFR激酶域的自身磷酸化,抑制EGFR的活性,进而阻断参与肿瘤生长与转移的EGFR信号转导通路,抑制肿瘤细胞生长,促进细胞凋亡。
1.1 EGFR酪氨酸激酶可逆抑制剂药物
20世纪90年代,阿斯利康研发的吉非替尼,是这类药物中最早进入临床和上市的口服EGFR酪氨酸激酶可逆抑制剂药物。
已上市的酪氨酸激酶可逆抑制剂代表性药物有吉非替尼、埃罗替尼、埃克替尼等,如图1所示。

图1 酪氨酸激酶可逆抑制剂Fig.1 Reversible inhibitors of tyrosine kinase
在吉非替尼上市后,相继上市了吉非替尼的“me-too”药物埃罗替尼、埃克替尼等。其中,浙江贝达药业有限公司通过对6-,7-位点修饰优化,成功上市了用于晚期NSCLC治疗的抗癌新药埃克替尼。
临床试验表明,埃克替尼的疗效及毒性与吉非替尼的疗效及毒性相似,但安全性更优。该药的成功研制,结束了我国TKIs类抗肿瘤药物全部进口的历史,打破了患者只能依赖于天价进口抗癌药的尴尬局面。
1.2 代表性EGFR酪氨酸激酶可逆抑制剂药物的合成
EGFR酪氨酸激酶可逆抑制剂(吉非替尼)合成路线主要有喹唑啉母核修饰、3,4-二甲氧基苯甲酸或3-羟基-4-甲氧基苯甲醛(酯)经脱保护、卤代、烷氧基化、缩合、重排等反应制备。
张华等人以3,4-二羟基苯甲醛为起始原料,经醛基缩合、氧化、侧链烷氧基修饰、重排环化制备4-(3-氯-4-氟苯氨基)-7-甲氧基-6-[3-(4-吗啉基)-丙氧基]喹啉(吉非替尼)的合成法更适宜工业化。
EGFR酪氨酸激酶可逆抑制剂类药物的开发是以喹唑啉骨架为母核,通过在母核上修饰合适的疏水基团,使分子与整个ATP结合域通过弱的、可逆性的作用力,如氢键、范德华力和疏水作用力等实现结合。这导致EGFR酪氨酸激酶可逆抑制剂类药物的选择性不够好、药效不够强烈和持久,同时还易引发耐药性。
2 EGFR酪氨酸激酶不可逆抑制剂
EGFR可逆抑制剂在临床上对NSCLC(非小细胞肺癌)患者,特别是对携带EGFR敏感型突变的患者的治疗取得了显著的治疗效果,但大部分患者在用药一段时间后(9~12个月),会出现获得性耐药的情况,超过半数的耐药原因是由于肿瘤细胞EGFR激酶结构域的新型突变,这个突变就是现在认识比较清晰的790位苏氨酸(Thr,简称T)点突变为甲硫氨酸(Met,简称M),一般称之为“T790M”。
2.1 EGFR酪氨酸激酶不可逆抑制剂药物
研究发现,当喹唑啉母核连接一个长度合适的亲电功能团,如各类不饱和酰胺结构侧链,这个亲电的功能团能与氨基酸残基共价结合时[EGFR激酶配体结合位点的半胱氨酸残基(C797)与迈克尔加成受体形成共价结合],使得药物牢固结合在靶点上与受体产生不可逆作用效果,大大提高了药物的抑制效率,这是EGFR不可逆抑制剂较为成熟的开发思路。
以此为基础,上市了EGFR酪氨酸激酶不可逆抑制剂,如阿法替尼、达克替尼、卡奈替尼,如图2所示。
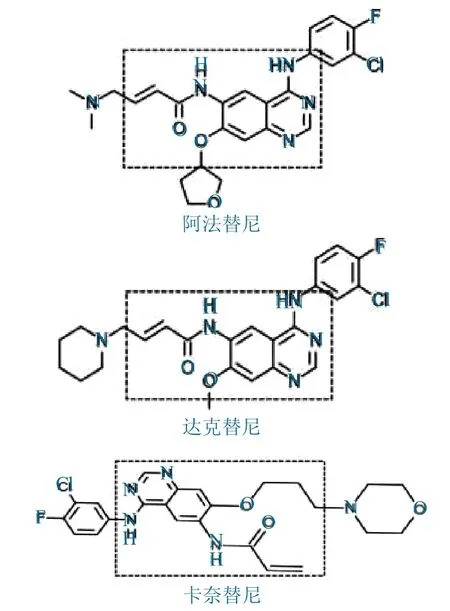
图2 酪氨酸激酶不可逆抑制剂Fig.2 Irreversible inhibitor of tyrosine kinase
2.2 代表性EGFR酪氨酸激酶不可逆抑制剂药物合成
阿法替尼是第1个上市的不可逆EGFR TKIs药物。阿法替尼主要有以下2种较为适宜的合成方案。
(1)从4-氯-2-氨基苯甲酸为起始原料,经环合、硝化、氯化、取代、醚化生成关键中间体N-4-[(3-氯-4-氟苯基)氨基]-6-硝基-7-[[(3S)-四氢-3-呋喃基]氧基]-喹唑啉,关键中间体再经氢化还原、缩合、Wittig-Horner反应及成盐,得到目标产物马来酸阿法替尼。(2)从中间体4-氯-6-氨基-7-羟基喹唑啉为原料,经醚化生成关键中间体4-氯-6-氨基-7-[[(S)-四氢呋喃-3-基]氧基]-喹唑啉后,再经氢化、酰胺化、取代、成盐得到马来酸阿法替尼。
3 喹唑啉类EGFR酪氨酸激酶抑制剂药物设计
上述喹唑啉类EGFR酪氨酸激酶抑制剂药物已经救助了上百万的肺癌患者,但在治疗一定时间后,都会发生耐药突变而丧失治疗效果,如T790M突变型、L858R合并T790M、Del19合并T790M双突变型和Del19/T790M/C797S及L858R/T790M/C797S三突变型等。这对医药工作者提出了针对这些突变开发特异性靶向EGFR酪氨酸激酶抑制剂的新要求。
综合已上市的芳胺基喹唑啉类EGFR酪氨酸激酶可逆抑制剂和EGFR酪氨酸激酶不可逆抑制剂两类药物的结构分析,喹唑啉类EGFR酪氨酸激酶抑制剂药物主要由3个六元环(A,B,C)和1个连接桥胺基构成,如图3所示。
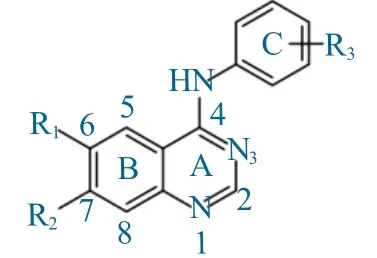
图3 芳胺基喹唑啉母核Fig.3 Arylamine quinazoline mother nucleus
通过对喹唑啉类EGFR酪氨酸激酶抑制剂药物的构效关系研究发现如下内容。
(1)环A的2个N原子与激酶间氢键的作用,对于药效活性至关重要。若A环1-位上的N原子与EGFR蛋白酶的Met769或Met793残基形成氢键,对激酶活性至关重要,如果1-位N原子被C原子替换,活性将降低3 700多倍。
(2)3-位上的N原子与Thr766形成氢键,如果3-N被C原子替换,活性将降低200多倍。
(3)B环对活性也有显著影响,8-位H被取代会因空间位阻阻止配体与受体之间的相互作用,对活性极其不利。
(4)6,7-位通常处于水相介质中,是优化化合物物化性质改善其药动学性质的重要修饰位点,提高此部位取代基团的亲水性,或采用能形成阳离子的基团,会提高TKIs的活性和增加生物的利用度。
(5)C环占据了EGFR-TKs的疏水空腔,影响着化合物的亲和力和选择性,特别是3,4-位以卤素等小基团取代时可显著提高活性。
(6)作为A环和C环连接桥梁的胺基最好裸露,该位置如被取代,将显著降低TKIs的活性。
生物构效与药效的关系研究表明,对喹唑啉母环的不同位置引入不同修饰基团,可以实现对药物活性的控制,这为新型EGFR酪氨酸激酶抑制剂药物开发提供了理论支持。
Daniel等人对EGFR不可逆抑制剂克服T790M突变耐药机制进行了研究,发现当喹唑啉母核6-位带有不饱和酰胺侧链时,能对EGFR-T790M表现出良好的抑制活性,在母核N(3)原子修饰后,会表现出对L858R/T790M变异体的抑制活性。
Carmi等人在母核6-位引入环氧酰胺基和苯氧乙酰氨基后,发现可以抑制EGFR的自磷酸化,并可以诱导L858R/T790M突变的NSCLC细胞凋亡。
韩美药品研发的波齐替尼Poziotinib(HM781-36B),通过对喹唑啉母核4-,6-位侧链修饰,实现了对EGFR外显子20插入突变的NSCLC的治疗。
薛子溪等人将喹唑啉与芳基哌嗪结构结合,设计了可能具有双靶点抗肿瘤小分子化合物喹唑啉-芳基哌嗪化合物。
孟祥川等人设计合成了一系列2-甲硫基-芳胺喹唑啉衍生物,实验显示部分化合物具有较好的抗肿瘤活性。
药物的复杂性远远不是上述的定向设计这么简单,但这些研究为开发更加高效、低毒、抗耐药的特异性靶向喹唑啉EGFR酪氨酸激酶抑制剂药物提供了思路。
4 结 语
喹唑啉类抗癌药物在治疗非小细胞肺癌、胰腺癌、乳腺癌等多种癌症疾病中,表现出了良好的治疗效果。本文对当前喹唑啉类表皮生长因子受体酪氨酸激酶抑制剂药物的发展历程、代表药物的合成方法、药物构效关系进行了综述。
目前,喹唑啉类抗癌药物已经成为治疗各种癌症疾病的一线用药。在药物构效关系指导下,结合现有研究成果和喹唑啉母核结构修饰位点,运用活性拼接理论,将对开发更加高效、低毒、抗耐药的特异性靶向喹唑啉类药物具有重要意义。
总之,喹唑啉类药物开发从理论研究和应用的角度来看,都具有很好的发展前景。
猜你喜欢
电子乐园·上旬刊(2022年5期)2022-04-09 22:25:58
天津医科大学学报(2021年3期)2021-07-21 09:03:46
世界科学技术-中医药现代化(2021年12期)2021-04-19 12:31:40
天然产物研究与开发(2018年8期)2018-09-10 05:48:18
天然产物研究与开发(2018年1期)2018-02-02 07:21:22
中成药(2018年1期)2018-02-02 07:19:57
中国塑料(2016年7期)2016-04-16 05:25:52
中国医药生物技术(2015年4期)2015-12-26 08:26:36
中国药业(2014年17期)2014-05-26 09:08:05
吉林大学学报(医学版)(2014年2期)2014-02-27 06:48:24
