
基于GEO数据库筛选狼疮性肾炎的关键基因和信号通路
2021-07-02 09:28谢裕赛王玉柱
上海交通大学学报(医学版) 2021年6期
刘 音,杨 涛,谢裕赛,王玉柱
1.北京市海淀医院肾内科,北京100080;2.中国医科大学基础医学院病理学教研室,沈阳110122
系统性红斑狼疮(systemic lupus erythematosus,SLE)是一种慢性系统性自身免疫性疾病,其特征是自身抗体产生、补体激活和免疫复合物沉积。每年全球范围内SLE 的发生率在0.003%~0.232%[1]。狼疮性肾炎(lupus nephritis,LN)是SLE患者中最常见、最严重的器官表现之一,其特征通常是血尿、蛋白尿和肾小球滤过率受损[2]。约50%的SLE 患者会发展为临床特征明显的肾脏疾病,其中11%的患者在病程5年时会发展为终末期肾脏疾病(end-stage renal disease,ESRD)[3-4]。LN 是导致SLE 患者发展至ESRD 和死亡的重要原因。LN 的初始和后续治疗主要由免疫抑制剂和糖皮质激素组成,这意味着几乎没有有效且特异性的疗法。因此,研究LN 涉及的分子机制对于LN的临床治疗具有重要意义。
LN 的病理生理机制复杂。在LN 患者的不同肾小球区室中,免疫复合物的形成、先天性免疫信号途径的激活、免疫细胞的渗透和促炎症介质可通过多种途径损害肾小球细胞[5]。尽管许多研究已经确定了LN 的某些病理机制,但其发病机制仍不清楚。
在基因组水平上追踪LN 的生物学变化是一种值得关注的策略。近年来,许多学者已经进行了基因测序技术与生物信息学分析相结合的研究,以鉴定可能与预后生物标志物有关的疾病相关基因,并在未来将其开发为治疗靶标。生物信息学分析可以在极短的时间内处理大量的样品,并提供有关疾病的有价值的信息,而且与SLE密切相关的几个基因已经确定,可以驱动研究的创新[6-7]。但是,很少有研究利用生物信息学分析来鉴定和分析LN背景下的肾脏组织。
尽管LN 会影响肾脏的所有组成部分,但肾小球是最适合研究的组织,并且与疾病的发病机制和治疗密切相关。本研究选择GEO 数据库中的肾小球组织数据集GSE32591 研究LN。应用R 语言limma 包获取GSE32591数据集中的差异表达基因(differentially expressed genes,DEGs)。使用DAVID 数据库来分析这些DEGs 的GO(Gene Ontology)注释信息,包括生物过程、细胞组分、分子功能和KEGG (Kyoto Encyclopedia of Genes and Genomes) 通路。构建了蛋白质-蛋白质相互作用(protein-protein interaction, PPI) 网络, 并应用Cytoscape 中MCODE 及Cytohubba 插件分析DEGs,鉴定并验证了与LN 相关的10 个枢纽基因(hub genes)。最终应用GSE99339 数据集验证了枢纽基因的显著差异表达,以期为LN的诊断和治疗提供新的思路。
1 资料与方法
1.1 数据来源及筛选
在GEO 公共数据库(http://www.ncbi.nlm.nih.gov/geo)[8]中以“lupus nephritis”及“glomeruli”作为关键词检索,选择GSE32591 数据集作为分析数据集,GSE99339 数据集作为枢纽基因验证数据集。下载GSE32591和GSE99339数据集矩阵数据及平台注释信息。GSE32591 数据集基于GPL14663 平台,其中包含32 例LN 患者和14 例正常供体对照的肾小球组织基因表达信息;GSE99339 数据集基于GPL19184 平台,含有30 例LN 患者和8 例肾脏肿瘤切除对照的肾小球组织基因表达信息。
1.2 筛选DEGs
应用R 软件(4.0.2 版) limma 包标准化数据及筛选差异基因[9],以|log FC|>1 和校正后P<0.05 为阈值筛选差异基因。分别运用R 语言ggpubr 和pheatmap 包对差异基因绘制火山图及热图。
1.3 DEGs的功能富集分析
使用GO 分析确定枢纽基因的生物过程、细胞成分和分子功能属性[10],通过KEGG 数据库确定其通路功能[11]。DAVID(https://david.ncifcrf.gov/) 是进行基因富集分析的常用数据库[12]。应用DAVID 在线工具对筛选的差异表达枢纽基因进行GO和KEGG分析,P<0.05为差异具有统计学意义,使用R语言ggplot包绘制柱状图及气泡图。
1.4 构建PPI网络及筛选枢纽基因
STRING 数据库常用来构建PPI 网络[13]。将DEGs 输入STRING 在线工具中,筛选combined score>0.9 的互作蛋白。将得到的PPI 结果导入Cytoscape 软件中,通过分子复合物检测(molecular complex detection,MCODE),以degree cutoff=2、node score cutoff=0.2、k-core=2 和max.depth = 100 为标准,筛选出PPI 中最显著模块[14]。在最显著模块中应用Cytohubba 插件,按degree 算法计算出评分位于前10位的枢纽基因[15]。
1.5 枢纽基因的验证
为了进一步验证正常组织和LN 组织中上述枢纽基因的表达差异,应用GEO 数据集GSE99339 的数据,分析枢纽基因的表达水平。
2 结果
2.1 DEGs的筛选
应用limma 包对GSE32591 数据集进行数据矫正及差异分析,以|log FC|>1 和校正后P<0.05 为阈值,获得了367个DEGs,包括253个上调基因及114个下调基因。使用ggpubr包绘制DEGs的火山图,按照校正后P值对显著DEGs进行排序;应用pheatmap包绘制出前20个DEGs的表达热图。结果显示,IFI27、MX1、ISG20等基因在LN中显著高表达(图1)。
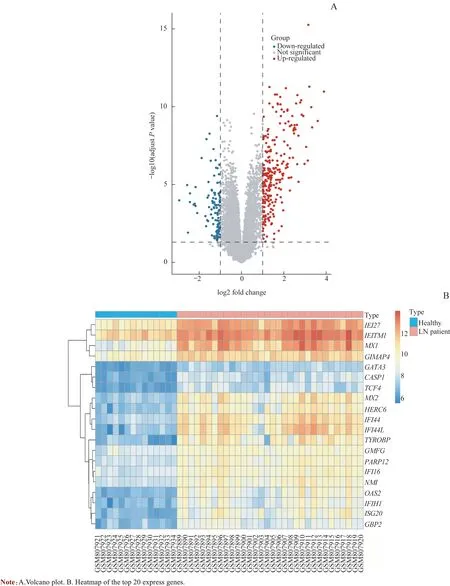
图1 DEGs的鉴定Fig 1 Identification of DEGs
2.2 DEGs的GO分析和KEGG信号通路分析
应用DAVID 软件对DEGs 进行GO 富集分析和KEGG信号通路富集分析。GO 富集分析显示这些DEGs 主要参与病毒反应应答、对病毒的防御反应,并且参与Ⅰ型干扰素信号通路、活化中性粒细胞以及负调控病毒生命周期等生物过程;主要细胞成分位于质膜外侧、囊腔及胶原纤维(含细胞外基质);主要分子功能与Toll 样受体结合、有机酸结合、补体结合及免疫受体活性等相关(图2A)。KEGG 通路分析显示,这些差异基因主要参与甲型流感、结核病、EB 病毒感染、金黄色葡萄球菌感染和补体途径等信号通路(图2B)。
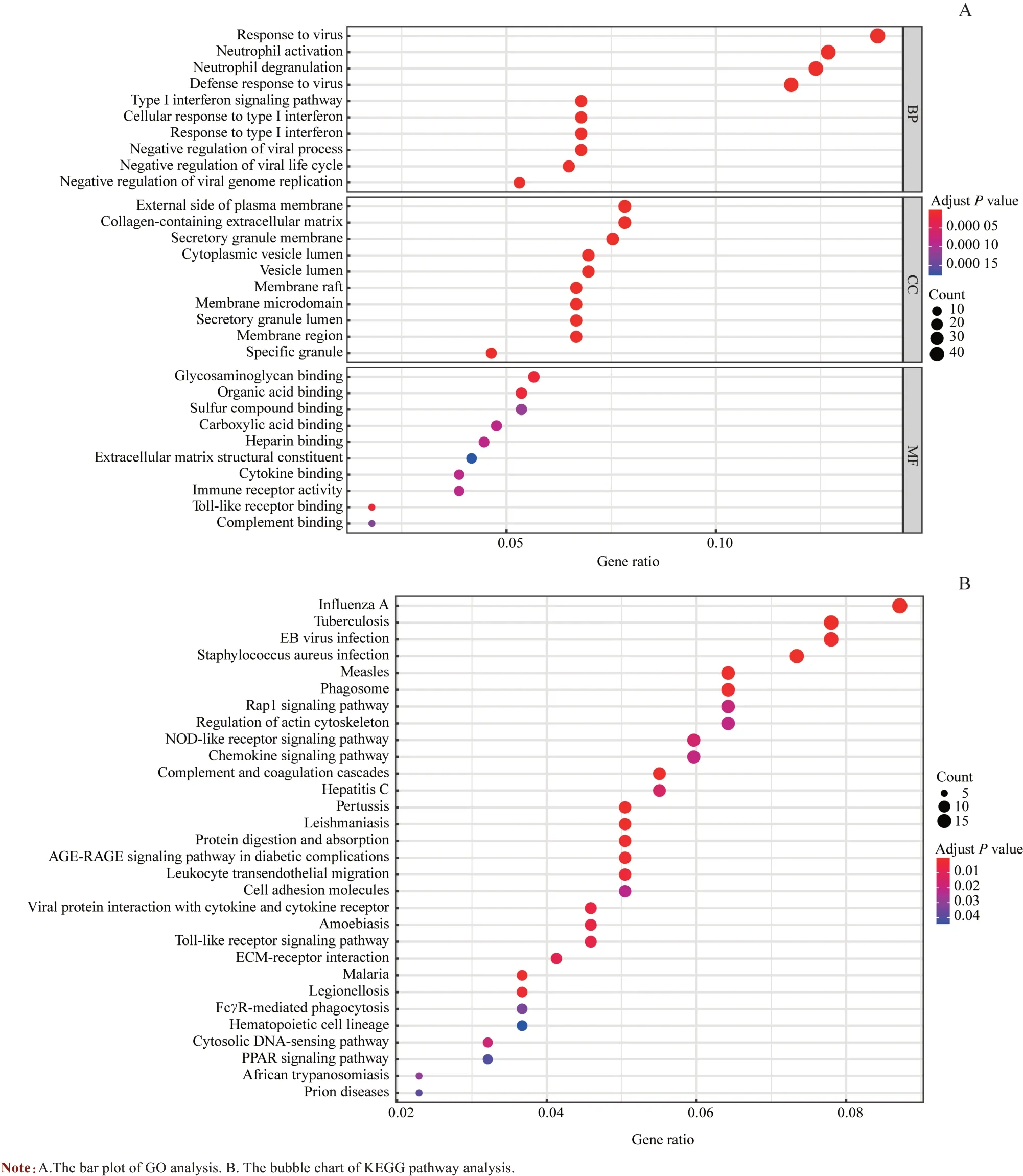
图2 DEGs的GO分析和KEGG通路分析Fig 2 GO analysis and KEGG pathway analysis of DEGs
2.3 DEGs编码蛋白的PPI网络及枢纽基因的筛选
通过STRING 在线工具和Cytoscape 软件对367 个DEGs 进行PPI 网络分析,筛选combined score >0.9 的互作蛋白对。在Cytoscape中得到206个节点,831条相互作用的网络图(图3A)。在PPI 网络中,利用Cytoscape 内的MCODE 插件, 以 degree cutoff = 2、 node score cutoff = 0.2、k-core = 2 和max.depth = 100 为标准,筛选出最显著模块。最显著模块包括22 个重要节点及227 条相互作用(图3B)。对最显著模块运用cytohubba 插件,以degree 算法计算出总分排名前10 位的枢纽基因,分别是IFI6、IFI27、IFIT1、IFIT2、IFIT3、IFITM3、ISG15、ISG20、MX1、SAMHD1(图3C,表1)。
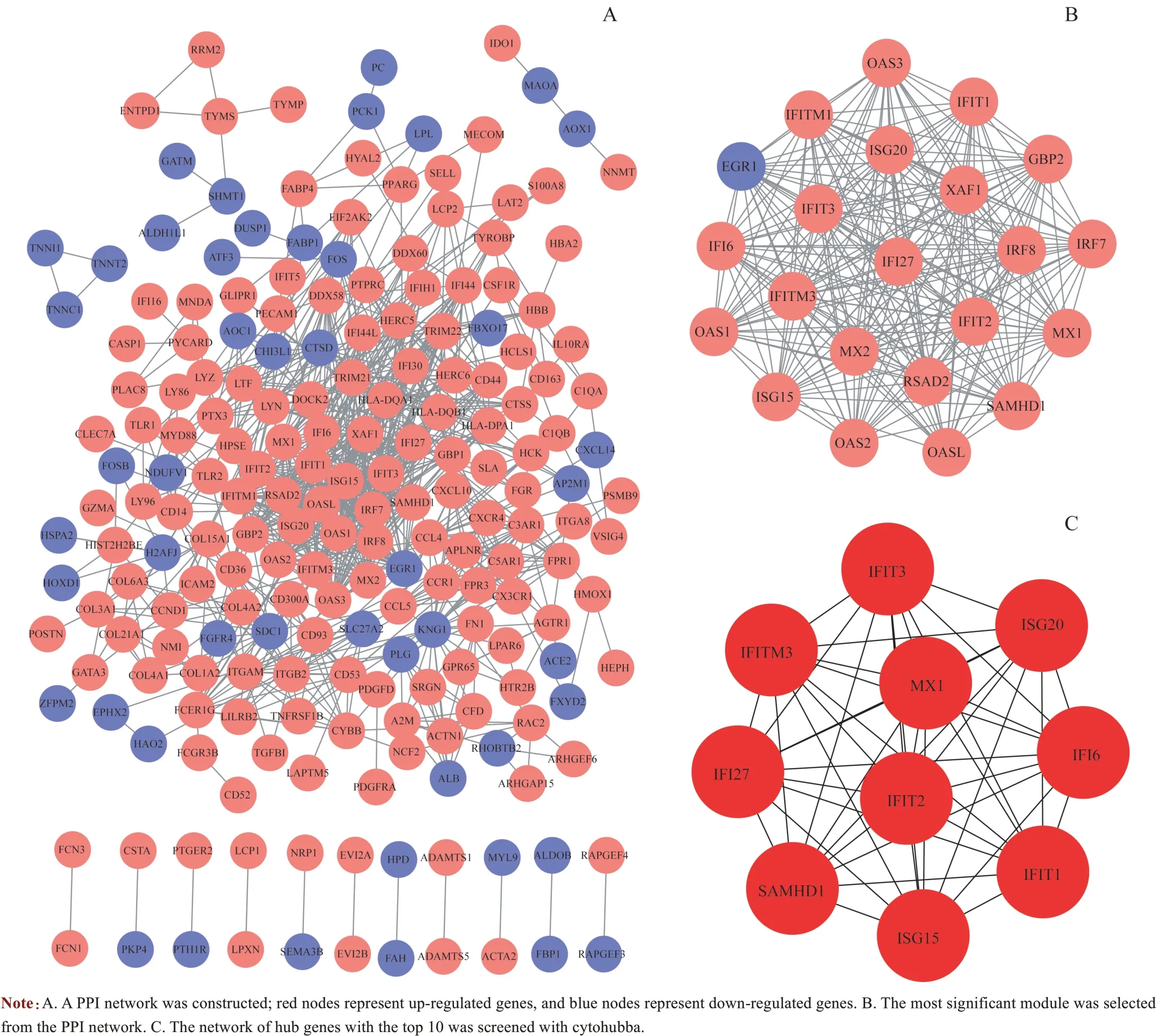
图3通过STRING和MCODE方法筛选枢纽基因Fig 3 Identification of hub genes from the DEGs by STRING and MCODE
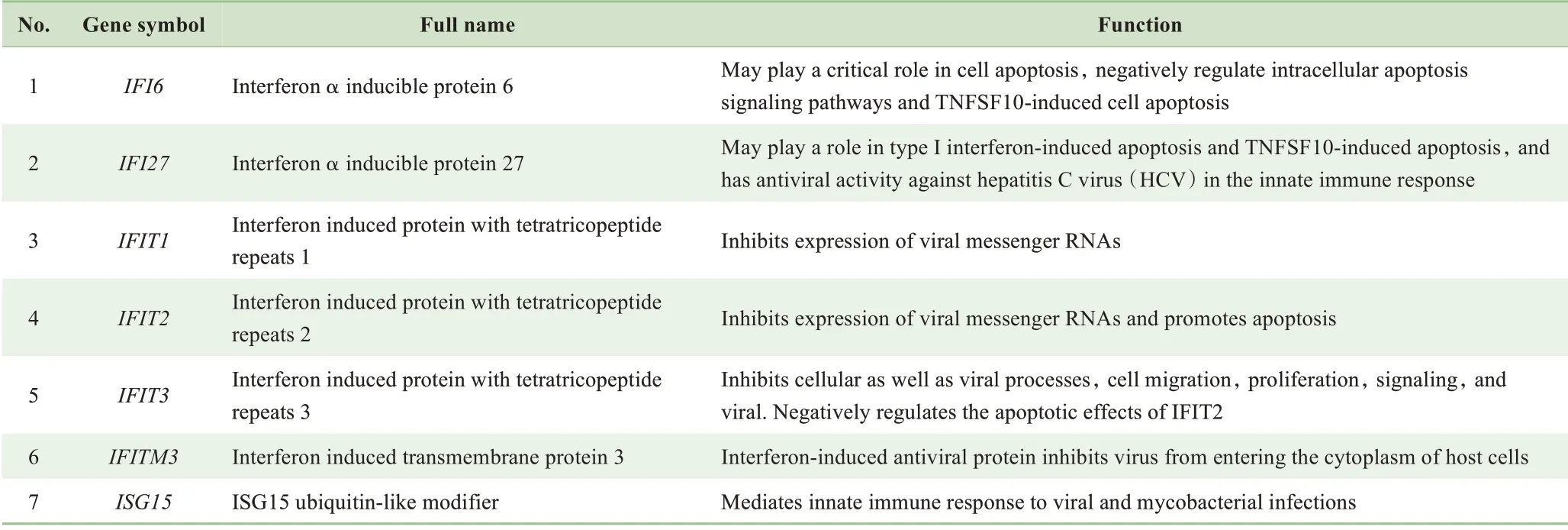
表1 10个枢纽基因的功能Tab 1 Functions of 10 hub genes
Continued Tab
2.4 枢纽基因的验证
在GEO 数据库检索其他LN 数据集。最终选择GSE99339 数据集,分析LN 与正常对照之间这些枢纽基因的差异表达水平(图4)。在GSE99339数据集中,除了缺失IFITM3的探针信息,上述枢纽基因均明显上调。说明了枢纽基因筛选结果的可靠性。
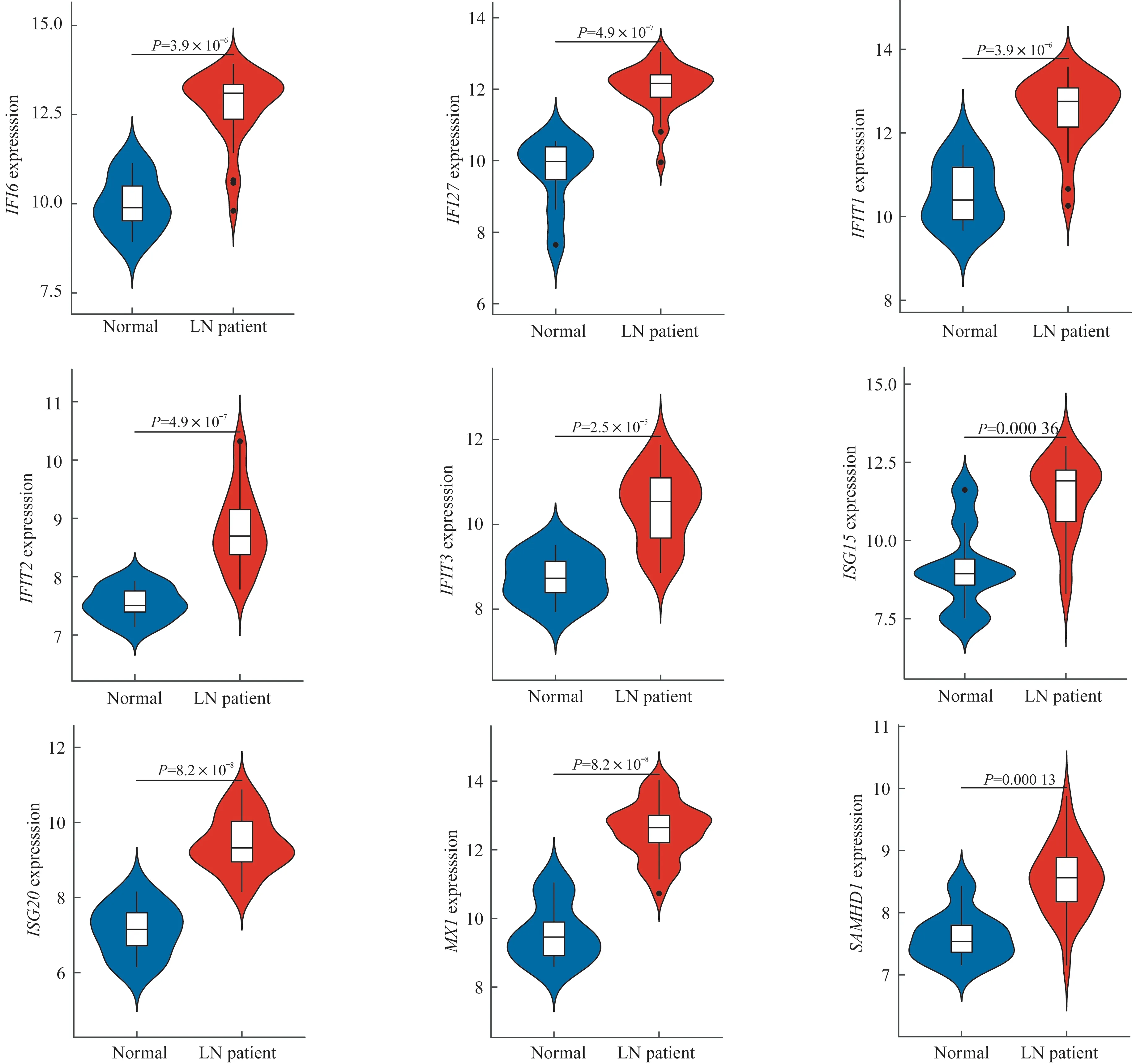
图4 在GSE99339数据集中验证枢纽基因在LN与正常组织之间的表达差异Fig 4 Differentially expressed levels of the hub genes between LN and normal in GSE99339 datasets
3 讨论
随着生物信息学的发展,人们对寻找各种疾病枢纽基因的关注日益增加,收集到的疾病枢纽基因信息可为治疗疾病提供新的手段。我们利用基于GPL14663平台的数据集GSE32591,对32 例LN 患者的肾小球组织基因表达数据进行分析,筛选DEGs。最终筛选出367 个DEGs,其中包括253 个上调基因和114 个下调基因;再运用ggpubr 和pheatmap 包绘制火山图及差异表达显著的前20个DEGs 的表达热图。热图结果显示,IFI27、MX1、ISG20等基因在LN 中显著高表达,提示这些基因可能参与LN的疾病进展。
对DEGs的功能富集分析显示,免疫应答、感染和干扰素相关基因参与了LN 的发病过程。GO 分析和KEGG途径富集分析表明,DEGs 在病毒防御反应、质膜外侧、甲型流感、结核病、EB 病毒感染和补体途径等方面显著富集,提示了LN 疾病进展中免疫活动的增强。本研究应用Cytoscape 中MCODE 及Cytohubba 插件分析这些DEGs,鉴定了与LN 相关的最显著模块及10 个枢纽基因(IFI6、IFI27、IFIT1、IFIT2、IFIT3、IFITM3、ISG15、ISG20、MX1和SAMHD1)。最终,应用其他GEO 数据集(GSE99339)的数据信息,验证了上述枢纽基因显著表达的可靠性。
在枢纽基因中,干扰素家族蛋白的表达显著上调,这也验证了之前的研究结果[16-17]。干扰素是一类信号蛋白,分为Ⅰ型、Ⅱ型和Ⅲ型;其中Ⅰ型干扰素是其最大的家族[18]。GO 分析结果也表明,Ⅰ型干扰素信号通路可能与SLE 的发病机制密切相关。SLE 的大多数枢纽基因都是Ⅰ型干扰素诱导型基因。SLE 患者显示出较高水平的Ⅰ型干扰素及Ⅰ型干扰素诱导型基因,这些基因在SLE 发病机制中起主要作用[19-20]。干扰素诱导的四肽重复序列1(IFIT1)蛋白质也属于Ⅰ型干扰素家族,在大多数SLE 人群中IFIT1mRNA 的表达明显上调[21]。IFIT1是首个鉴定为主要由干扰素-α/β诱导的细胞质和线粒体干扰素刺激基因[22]。IFIT1可调节病毒复制以及翻译、增殖、凋亡和信号转导等细胞过程[23],可能与Rho/Rac鸟嘌呤核苷酸交换因子发生相互作用,从而参与SLE免疫反应。
在本研究中,我们使用系统的生物信息学方法来分析LN 中肾小球组织表达信息。研究存在一些局限性,如GEO数据库仅提供了少量LN数据集。在后续研究中,我们将进一步收集临床数据,以作更深入的探索。
总之,本研究使用GEO 数据库筛选LN 肾小球组织的367 个DEGs 及10 个枢纽基因,其可能是LN 潜在的生物标志物。生物标志物的揭示,有助于提高临床诊断水平和开发个性化治疗方法。
参·考·文·献
[1] Rees F, Doherty M, Grainge MJ, et al. The worldwide incidence and prevalence of systemic lupus erythematosus: a systematic review of epidemiological studies[J]. Rheumatology (Oxford), 2017, 56(11): 1945-1961.
[2] Yu F,Haas M,Glassock R,et al. Redefining lupus nephritis:clinical implications of pathophysiologic subtypes[J]. Nat Rev Nephrol,2017,13(8):483-495.
[3] Almaani S, Meara A, Rovin BH. Update on lupus nephritis[J]. Clin J Am Soc Nephrol,2017,12(5):825-835.
[4] Tektonidou MG, Dasgupta A, Ward MM. Risk of end-stage renal disease in patients with lupus nephritis, 1971-2015: a systematic review and Bayesian meta-analysis[J]. Arthritis Rheumatol,2016,68(6):1432-1441.
[5] Devarapu SK, Lorenz G, Kulkarni OP, et al. Cellular and molecular mechanisms of autoimmunity and lupus nephritis[J]. Int Rev Cell Mol Biol,2017,332:43-154.
[6] Bing PF, Xia W, Wang L, et al. Common marker genes identified from various sample types for systemic lupus erythematosus[J]. PLoS One, 2016,11(6):e0156234.
[7] Coit P, Jeffries M, Altorok N, et al. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferonregulated genes in naïve CD4+T cells from lupus patients[J]. J Autoimmun,2013,43:78-84.
[8] Clough E, Barrett T. The gene expression omnibus database[J]. Methods Mol Biol Clifton N J,2016,1418:93-110.
[9] Piao J,Sun J,Yang Y,et al. Target gene screening and evaluation of prognostic values in non-small cell lung cancers by bioinformatics analysis[J]. Gene,2018,647:306-311.
[10] Gene Ontology Consortium. Gene Ontology Consortium: going forward[J].Nucleic Acids Res,2015,43(database issue):D1049-D1056.
[11] Du JL,Yuan ZF, Ma ZW, et al. KEGG-PATH: Kyoto encyclopedia of genes and genomes-based pathway analysis using a path analysis model [J]. Mol Biosyst,2014,10(9):2441-2447.
[12] Dennis G, JR., Sherman BT, Hosack DA, et al. DAVID: Database for annotation, visualization, and integrated discovery[J]. Genome Biol, 2003,4(5):P3.
[13] Kumar MS, Adki KM. Marine natural products for multi-targeted cancer treatment:a future insight[J]. Biomed Pharmacother,2018,105:233-245.
[14] Wang J, Zhong J, Chen G, et al. ClusterViz: a cytoscape APP for cluster analysis of biological network[J]. IEEE/ACM Trans Comput Biol Bioinform,2015,12(4):815-822.
[15] Chin CH,Chen SH,Wu HH,et al. cytoHubba:identifying hub objects and subnetworks from complex interactome[J]. BMC Syst Biol,2014,8(Suppl 4):S11.
[16] Yang H, Li H.CD36identified by weighted gene co-expression network analysis as a hub candidate gene in lupus nephritis[J]. PeerJ, 2019, 7:e7722.
[17] Shu B, Fang Y, He W, et al. Identification of macrophage-related candidate genes in lupus nephritis using bioinformatics analysis[J]. Cell Signal, 2018,46:43-51.
[18] Reder AT, Feng X. Aberrant type I interferon regulation in autoimmunity:opposite directions in MS and SLE,shaped by evolution and body ecology[J].Front Immunol,2013,4:281.
[19] Bezalel S, Guri KM, Elbirt D, et al. Type I interferon signature in systemic lupus erythematosus[J]. Isr Med Assoc J,2014,16(4):246-249.
[20] Chen JY, Wang CM, Chen TD, et al. Interferon-λ3/4 genetic variants and interferon-λ3 serum levels are biomarkers of lupus nephritis and disease activity in Taiwanese[J]. Arthritis Res Ther,2018,20(1):193.
[21] Becker AM, Dao KH, Han BK, et al. SLE peripheral blood B cell, T cell and myeloid cell transcriptomes display unique profiles and each subset contributes to the interferon signature[J]. PLoS One,2013,8(6):e67003.
[22] Reynaud JM,Kim DY,Atasheva S,et al. IFIT1 differentially interferes with translation and replication of alphavirus genomes and promotes induction of type I interferon[J]. PLoS Pathog,2015,11(4):e1004863.
[23] Zhang L, Wang B, Li L, et al. Antiviral effects of IFIT1 in human cytomegalovirus-infected fetal astrocytes[J]. J Med Virol, 2017, 89(4):672-684.
猜你喜欢
承德医学院学报(2022年2期)2022-05-23
天津医科大学学报(2021年4期)2021-08-21
电子技术与软件工程(2021年4期)2021-06-16
保健与生活(2021年8期)2021-04-19
农民致富之友(2020年18期)2020-06-19
电子制作(2019年22期)2020-01-14
火力与指挥控制(2019年5期)2019-06-13
软件(2016年6期)2017-02-06
妇女生活(2016年3期)2016-03-11
中国动物保健(2015年6期)2015-10-21
