
司美替尼下调KRAS G12V 突变型非小细胞肺癌细胞PD-L1水平的探索性研究
2021-07-02 09:28马韵芳潘丽娜高蓓莉胡家安徐志红
上海交通大学学报(医学版) 2021年6期
马韵芳,潘丽娜,李 圳,高蓓莉,胡家安#,徐志红#
1.上海交通大学医学院附属瑞金医院老年病科,上海200025;2.上海交通大学医学院附属瑞金医院呼吸与危重症科,上海200025
非小细胞肺癌(non-small cell lung cancer,NSCLC)占所有肺癌的80%~85%,是全球癌症死亡的首要病因[1]。Kirsten 大鼠肉瘤病毒癌基因(Kirsten rat sarcoma viral oncogene,KRAS)是NSCLC患者中最常见的致癌驱动突变基因之一[2-3],15%~20%的NSCLC患者携带有KRAS基因突变。KRAS突变主要位于12号染色体的外显子12、13和61,其中外显子12 突变约占NSCLC 所有KRAS突变的90%[3]。KRAS突变已被认为是NSCLC预后不良的标志,预示患者总生存期的缩短和肿瘤复发风险的升高[4]。KRAS突变的NSCLC患者并不能从铂类化学治疗方案中获得总生存期的延长,对表皮生长因子受体(epidermal growth factor receptor,EGFR) 酪氨酸激酶抑制剂(tyrosine kinase inhibitors,TKIs)的治疗反应也很差。目前,KRAS突变型NSCLC患者迫切需要更有效的治疗策略。
免疫检查点抗体通过靶向程序性死亡蛋白配体1(programmed cell death ligand 1,PD-L1)或程序性死亡蛋白1 (programmed cell death 1,PD-1) 阻断PD-1 通路,在以KRAS/RAF突变为常见驱动事件的多种恶性肿瘤中(包括NSCLC)显示出良好的临床疗效[5]。然而,关于抗PD-1 抗体(纳武单抗和帕姆单抗)的临床研究指出,在非选择性NSCLC 患者中,PD-1 抗体的应答率仅为20%[6]。多项临床试验表明抗PD-1/PD-L1 药物的疗效与NSCLC 患者肿瘤中PD-L1 的阳性率密切相关,PD-L1表达越高则疗效越佳[6-11]。对肿瘤细胞PD-L1 阳性率≥50%的NSCLC 患者使用帕姆单抗治疗,其应答率和无进展生存期均有所提高[7]。在该患者队列中,KRAS突变型比KRAS野生型肿瘤PD-L1 阳性率更高。Kim 等[12]所做的meta 分析显示,在KRAS突变的晚期NSCLC 患者中,免疫检查点抑制剂与多西他赛相比,提高了患者的总生存率。这些证据表明KRAS突变状态可能是免疫治疗生存获益的潜在生物标志物。Lan 等[13]的研究观察到,PD-L1 的表达与KRAS基因突变存在显著的相关性(P=0.006)。然而,NSCLC 患者不同KRAS突变亚型与PD-L1 表达之间的相关性目前尚不清楚。本研究为进一步探索KRAS突变亚型与PD-L1 表达之间的关系,首先在NSCLC 细胞系中检测了KRAS的突变状态以及PD-L1mRNA 和蛋白的表达水平,并分析两者之间的关系。其次,采用免疫组织化学(免疫组化)方法进一步检测了77 例早期NSCLC 患者中PD-L1 的表达情况。然后,观察了KRAS突变状态对RAS 下游信号通路的影响。最后,探索了TKIs (RAS 下游通路信号分子的抑制剂) 对KRAS突变型NSCLC 细胞PD-L1 表达的调控作用,以探讨KRAS调控PD-L1 的分子机制。
1 材料与方法
1.1 细胞株和肿瘤样本
本研究使用的47 株NSCLC 细胞系,包括25 个KRAS突变株和22 个KRAS野生型(wild type,WT)细胞株,分别购自美国模式培养物集存库(ATCC)、日本理化学研究所(RIKEN)、日本研究生物资源(JCRB)细胞库和欧洲认证细胞培养物收藏中心(ECACC)。细胞株均采用含10%胎牛血清、青霉素(100 IU/mL) 和链霉素(100 μg/mL)的适宜培养基,在37 ℃、含5% CO2的加湿环境中培养。所有实验均使用指数生长期的细胞。
本研究共纳入2013—2017 年在上海交通大学医学院附属瑞金医院接受常规手术且经病理学检查确诊为早期NSCLC(Ⅰa~Ⅱb 期)的患者共77 例,均未接受术前治疗。根据国际抗癌联盟颁布的第8 版肺癌分期标准[14]对患者进行分类。这些患者的肿瘤标本按照标准程序经甲醛溶液固定后制成石蜡包埋组织。
本研究经上海交通大学医学院附属瑞金医院机构审查委员会和伦理委员会批准,所有患者均签署书面知情同意书。
1.2 主要试剂和仪器
TKIs 包括RAF 抑制剂达拉非尼、有丝分裂原活化蛋白激酶(mitogen activated-protein kinase,MEK)抑制剂司美替尼、磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)抑制剂GDC0941、蛋白激酶B(protein kinase B,PKB,又称为AKT)抑制剂MK2206、哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR) 抑制剂雷帕霉素、 核糖体S6 蛋白激酶(ribosomal S6 protein kinase,p70S6K)抑制剂LY2584702均购自MedChem Express公司。
胎牛血清购自美国Hyclone公司,青霉素和链霉素购自美国Life Technologies 公司,蛋白酶抑制剂购自瑞士Roche 公司, 聚偏氟乙烯(polyvinylidene fluoride,PVDF)转移膜、化学发光试剂购自美国Millipore 公司,EDTA 缓冲液(pH 9.0, G1203-250ML) 购自中国Servicebio 公司,RNeasy Mini 试剂盒购自德国QIAGEN公司,PrimeScript RT 试剂盒购自日本TaKaRa 公司,SYBR Green 试剂购自美国Life Technologies 公司,扩增引物购自上海赛百盛基因技术有限公司,DAB 显色试剂盒购自丹麦Dako公司。
AKT抗体(sc-8312)购自美国Santa Cruz公司,p-AKT抗体(S473,#4060)、细胞外调节蛋白激酶(extracellular regulated protein kinase,ERK)抗体(#4695)、p-ERK 抗体(T202/Y204, #9106)、 p-p70S6K 抗体(T389, #9206)购自美国Cell Signaling Technology公司,mTOR抗体(6585-1) 购自美国Epitomics 公司,p-mTOR 抗体(S2448,ab109268)、p70S6K 抗体(ab9366)、PD-L1 抗体(ab228462)购自英国Abcam 公司,甘油醛-3-磷酸脱氢 酶 (glyceraldehyde-3 phosphate dehydrogenase,GAPDH)抗体(080922)购自中国Abmart公司,辣根过氧化物酶(HRP)标记的山羊抗兔IgG(G1215-200T)购自中国Servicebio公司。
移液器、低温高速离心机(Eppendorf,德国),实时荧光定量PCR 仪(Roche,瑞士),ChemiScope 3500 化学发光成像仪(上海勤翔科学仪器有限公司),流式细胞仪FACStation(BD,美国)。
1.3 实时定量PCR
分别提取47 个NSCLC 细胞株的RNA,反转录合成cDNA,再以cDNA 为模板进行实时定量PCR(real-time quantitative PCR,qPCR),扩增PD-L1基因。正向引物为5′-CCGAAGTCATCTGGACAAGCA-3′,反向引物为5′-CTTCTCCTCTCTCTTGGAATTGGT-3′。PCR 扩增条件:94 ℃预变性4 min;94 ℃变性40 s,50~60 ℃退火40 s,72 ℃延伸40 s,共35个循环;72 ℃延伸6 min,4 ℃保存。
1.4 Western blotting
提取NSCLC 细胞的总蛋白,蛋白定量后进行十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)分离,然后将蛋白转移至PVDF 膜。经5%脱脂牛奶室温封闭2 h后,加入一抗(GAPDH 抗体、AKT 抗体、p-AKT 抗体、ERK 抗体、p-ERK 抗体、mTOR 抗体、p-mTOR 抗体、p70S6K抗体以及p-p70S6K抗体)稀释液,于摇床上室温孵育2 h。PBS 洗膜后二抗(1∶2 000)室温孵育1 h。滴加化学发光试剂显影,调整曝光,置于全自动化学发光分析仪中检测。图像灰度由Image J软件进行分析。
1.5 荧光活化细胞分选法
用胰蛋白酶消化细胞,用培养液洗涤细胞,然后在荧光活化细胞分选(fluorescence activated cell sorting,FACS)缓冲液中进行抗体染色。流式细胞仪检测PD-L1的表达,使用CytExpert 1.1软件进行分析。PD-L1相对表达量=PD-L1的平均荧光强度/同型对照的平均荧光强度。
1.6 免疫组化染色
将77 例NSCLC 肿瘤患者肺癌组织连续切片、脱蜡、水化后,置于EDTA 缓冲液(pH 9.0)中于微波炉内进行抗原修复。阻断内源性过氧化物酶处理后用3%牛血清白蛋白室温封闭30 min。加入PD-L1 单克隆抗体(1∶100稀释),于4 ℃冰箱孵育过夜。PBS 洗涤,加入HRP 标记的二抗(1∶2 000 稀释)室温下孵育50 min 后PBS 冲洗3次。最后,使用DAB 显色试剂盒显色,脱水封片,进行染色分析。
1.7 阳性细胞计数方法及结果判读
本次实验采用肿瘤细胞阳性比例分数(tumor proportion score,TPS)的计数方式(即只计数肿瘤细胞),阳性肿瘤细胞计数方法及结果判读标准参照PD-L1(22C3)专用检测试剂盒的判读标准及计分标准。判读标准:计数的肿瘤细胞数≥100 个;任何染色强度的膜染色(包括完整和/或不完整膜阳性)均为阳性,单纯胞质或胞核染色不能判为阳性染色;阳性结果记录为具体的百分比。TPS<1%为阴性,TPS介于1%~49%为低表达,TPS≥50%为高表达。所有染色结果均由2位病理科专家进行独立判定和评分。
1.8 统计学分析
采用SPSS 25.0 软件进行统计学分析。正态分布定量资料用±s表示,多组间比较采用方差分析,两两比较采用Dunnett-t检验;非正态分布定量资料2组间比较采用秩和检验。定性资料组间比较采用χ2检验。P<0.05 为差异有统计学意义。
2 结果
2.1 NSCLC 细胞株和肿瘤组织中PD-L1 表达与KRAS 突变状态有关
采用qPCR 法检测47 个NSCLC 细胞株PD-L1mRNA的表达情况,包括25 个KRAS突变株和22 个KRASWT 细胞株。如图1 所示,KRAS突变细胞株的PD-L1mRNA 表达水平显著高于KRASWT 细胞株(P=0.003),而EGFR、TP53不同突变状态,不同肺癌组织类型(腺癌与鳞状细胞癌)NSCLC 细胞株的PD-L1mRNA 表达水平差异均无统计学意义。此外,KRASG12V 突变、KRASG12C 突变及其他KRAS突变细胞株的PD-L1mRNA表达水平均显著高于KRASWT 细胞株(P值分别为0.000、 0.033、0.045),其中KRASG12V 突变株的PD-L1mRNA 表达水平最高。
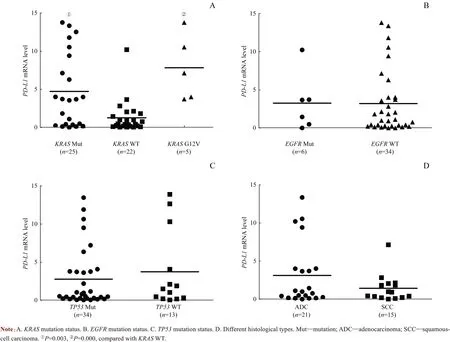
图1 NSCLC细胞株PD-L1 mRNA表达与KRAS、EGFR、TP53突变状态及肺癌组织学类型间的关系Fig 1 Correlations between PD-L1 mRNA expression and KRAS,EGFR,TP53 mutation status and histological types in the NSCLC cell lines
通过流式细胞术检测KRASWT 和KRAS突变型NSCLC 细胞株中PD-L1 的表达情况,结果(图2)显示KRAS突变株PD-L1 蛋白的表达约是KRASWT 株的23 倍(54.2±7.0vs2.4±0.4,P=0.000),其中KRASG12C 突变株的PD-L1蛋白表达水平最高,在不同突变亚型之间PD-L1蛋白表达差异无统计学意义。
此外,本研究通过免疫组化法检测了77例早期(Ⅰa~Ⅱb期)NSCLC患者肿瘤组织中PD-L1的表达情况(图3),包括10例WT型、15例G12A突变型、12例G12C突变型、21例G12D突变型和19例G12V突变型。结果显示,KRASG12D和G12V突变型中分别有28%和53%患者肿瘤组织PD-L1的TPS≥50%,显著高于KRASWT患者(表1)。
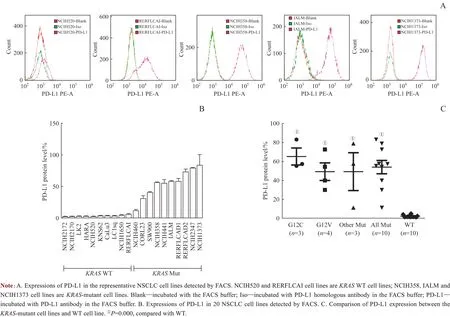
图2 FACS检测PD-L1在NSCLC细胞系中的表达情况Fig 2 Expression of PD-L1 in the NSCLC cell lines detected by FACS

图3 早期NSCLC患者肿瘤组织免疫组化染色Fig 3 Immunohistochemical staining of tumor tissues in the early NSCLC patients
2.2 KRAS 突变型NSCLC 细胞株中,p70S6K 被激活,而AKT、mTOR未被激活
KRAS是RAS 家族的成员之一,参与调控RAS/PI3K/AKT/mTOR/p70S6K 信号通路。因此,本研究检测了14个NSCLC 细胞株(包括4 个KRASWT 和10 个KRAS突变细胞株)中AKT、p-AKT、mTOR、p-mTOR、p70S6K和p-p70S6K 蛋白的表达。结果显示,NSCLCKRAS突变株和WT 株AKT 与mTOR 信号分子的表达无明显差异(图4A),且蛋白质半定量分析亦表明p-AKT/AKT 和p-mTOR/mTOR 在2 组间差异无统计学意义(图4B);而蛋白质半定量分析结果提示KRAS突变株中p-p70S6K/p70S6K比值显著增加(P=0.031)。
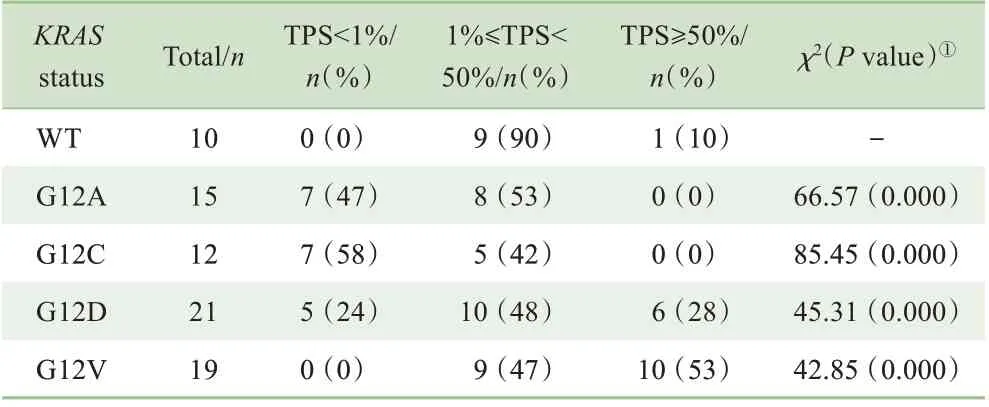
表1 不同KRAS状态的早期NSCLC患者肿瘤组织PD-L1表达情况Tab 1 Expression of PD-L1 in the tumor tissues of early NSCLC patients with different KRAS states
随后,用300 nmol/L和3 μmol/L LY2584702(p70S6K抑制剂)分别处理7 个NSCLC 细胞株(5 个KRAS突变株和2 个KRASWT 株)24 h 后,流式细胞术观察PD-L1 表达的变化;结果发现300 nmol/L LY2584702 可诱导KRASG12C(NCIH1373)细胞株PD-L1 表达上调(P=0.039),而3 μmol/L LY2584702 则可使KRASG12V (CORL23)细胞株PD-L1表达下降(P=0.001),而在其他5个细胞株LY2584702均未引起PD-L1的明显变化(图5)。
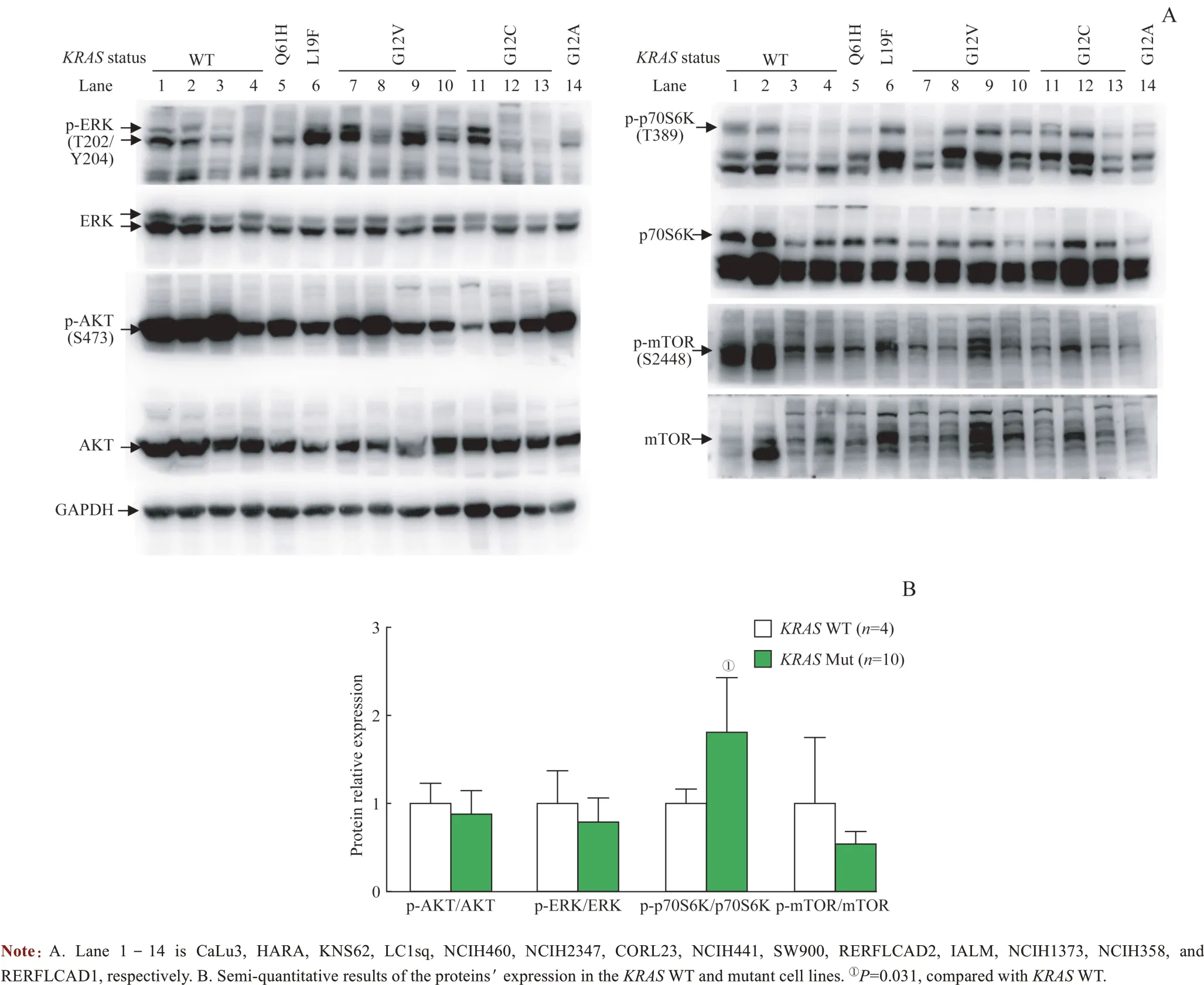
图4 Western blotting检测RAS下游信号分子的表达Fig 4 Expression of RAS downstream signaling molecules by Western blotting
用1 μmol/L GDC0941(PI3K 抑制剂)、0.5 μmol/L MK2206(AKT 抑制剂)和10 nmol/L 雷帕霉素(mTOR抑制剂)分别处理6 种KRAS突变株24 h,同样未观察到PD-L1蛋白水平的变化(图6A)。
2.3 在KRAS 突变型NSCLC 细胞株中,RAF 和MEK 对PD-L1的调控存在差异
KRAS不仅参与调控RAS/PI3K/AKT/mTOR/p70S6K信号通路,还参与调控RAS/RAF/MEK/ERK 信号通路。本研究检测了14 个NSCLC 细胞株(包括4 个KRASWT和10 个KRAS突变细胞株)中pERK、ERK 的蛋白水平。结果显示,NSCLCKRAS突变株和WT 株之间p-ERK/ERK比值差异无统计学意义(图4)。
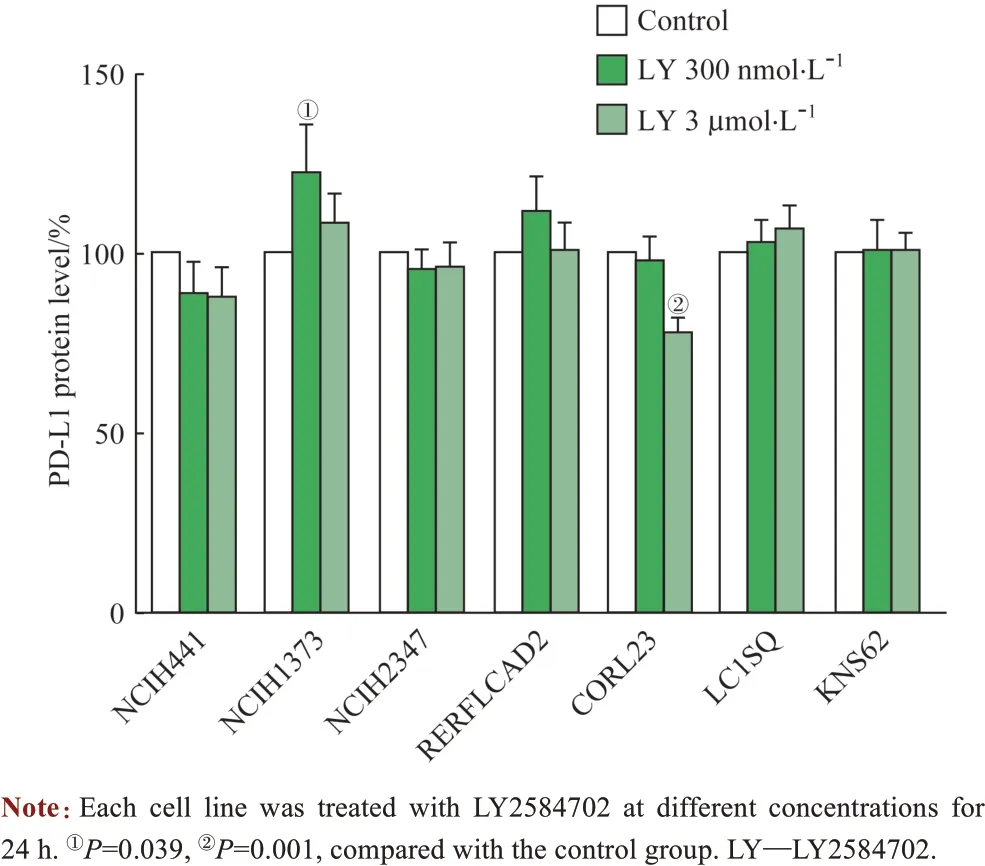
图5 FACS检测LY2584702对PD-L1表达的影响Fig 5 Influence of LY2584702 on the expression of PD-L1 by FACS
进一步用不同浓度达拉非尼(RAF 抑制剂)和司美替尼(MEK 抑制剂)分别处理6 种KRAS突变株24 h,通过流式细胞术检测PD-L1 蛋白的表达。结果发现,低浓度(5 nmol/L) 达拉非尼仅在1 个KRASG12V 突变株(CORL23)和1 个KRASL19F 突变株(NCIH2347)引起PD-L1 表达的显著升高(均P<0.05),且呈一定剂量依赖性(图6B)。而低浓度(0.1 μmol/L)司美替尼在3 个KRASG12V 突 变 株 (NCIH441、 CORL23 和RERFLCAD2) 和1 个KRASL19F 突变株(NCIH2347)引起PD-L1 表达的显著变化,但均表现为下调(均P<0.05),亦呈剂量依赖性(图6B)。在其余突变细胞株中,达拉非尼或司美替尼需达到较高浓度才能引起PD-L1 的升高或降低。
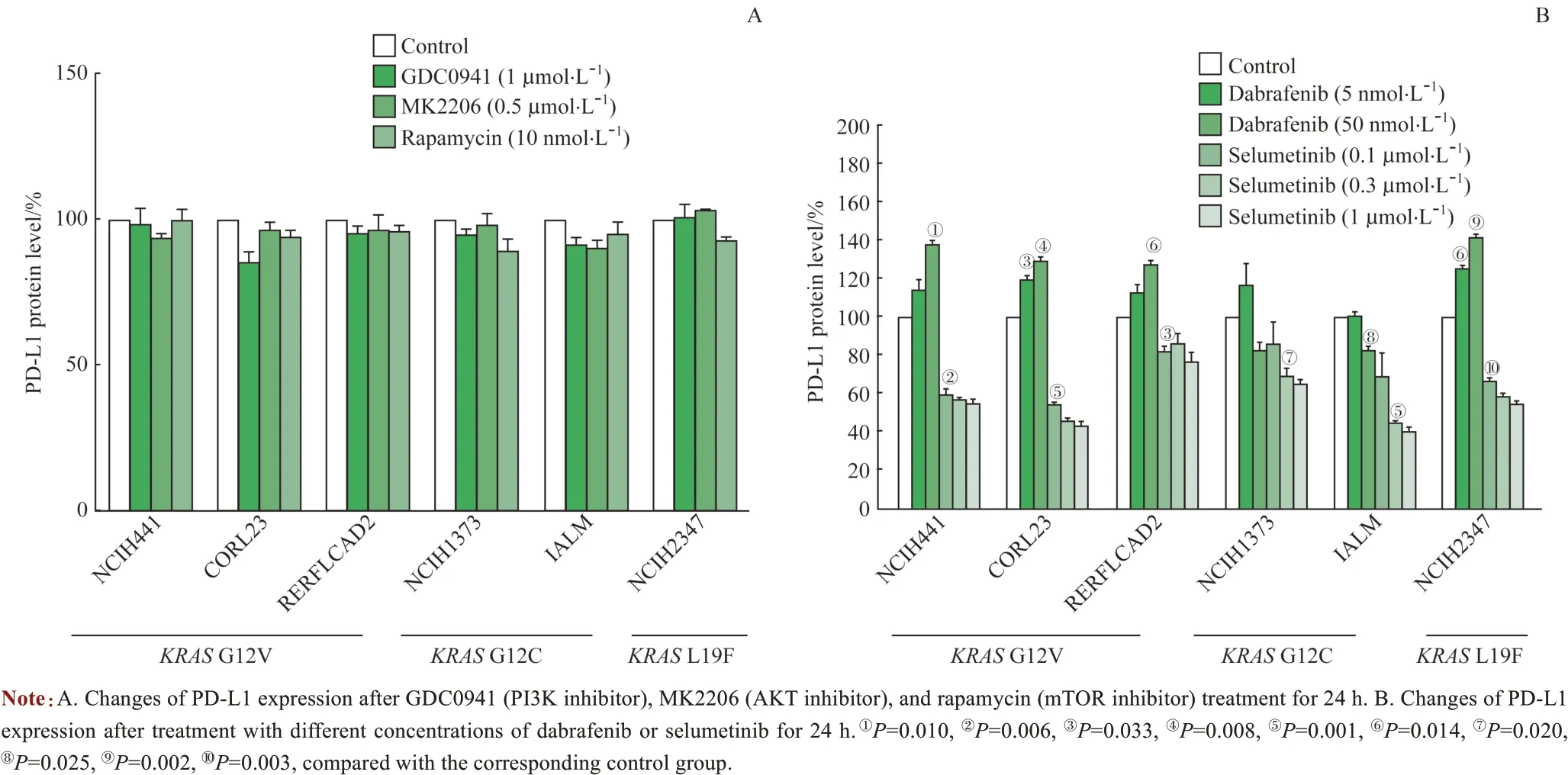
图6 FACS检测TKIs对KRAS突变NSCLC细胞株PD-L1表达的调控Fig 6 Regulation of TKIs on the expression of PD-L1 in KRAS-mutant NSCLC cell lines by FACS
3 讨论
在2020 版《非小细胞肺癌诊疗指南》中,多款免疫新药被推荐用于NSCLC的一、二线治疗,其中包括PD-1/PD-L1 抑制剂。多项临床试验[6-11]表明抗PD-1/PD-L1 药物的疗效与NSCLC 患者肿瘤中PD-L1 的表达水平密切相关:PD-L1 高表达(免疫组化TPS≥50%)的NSCLC 患者从PD-1 单抗治疗中获益最大,其客观缓解率、中位无进展生存期、总生存期显著好于TPS<50%的群体。
肺癌中PD-1/PD-L1 的调控是一个复杂的过程,与多个驱动基因及多条信号通路有关。前期研究[12]发现,肺癌驱动基因KRAS突变患者的肿瘤组织PD-L1 的表达较KRASWT 型高。但关于NSCLC 患者KRAS各突变亚型与PD-L1表达水平之间的关系,目前相关研究甚少,且结果不尽相同。
本研究发现,KRAS突变型NSCLC 细胞株PD-L1mRNA 和蛋白水平明显高于KRASWT 细胞株,结果与既往研究[14-16]相符。其中,KRASG12V 突变细胞株PD-L1的mRNA 表达水平最高,G12C 突变细胞株PD-L1 的蛋白表达水平最高,这2 个亚型共占NSCLC 所有KRAS突变的60%以上。我们进一步采用免疫组化方法检测了77 例早期NSCLC 患者中PD-L1 的表达情况,结果显示PD-L1在KRASG12D 和G12V 突变型中呈高表达,而在G12C 和G12A 突变患者中表达不高,其结果似乎与上述NSCLC细胞层面的结果不完全一致。考虑到大多数NSCLC 细胞株来源于晚期肿瘤组织,而免疫组化的标本为早期NSCLC 患者肿瘤组织,故两者之间存在差异。但无论是细胞层面还是组织层面,我们发现KRASG12V 突变型的PD-L1 表达均较高。我们由此猜测,KRASG12V 突变的NSCLC患者接受抗PD-1/PD-L1免疫治疗的获益率可能更高。阐明NSCLC 中KRAS突变状态与PD-L1 表达之间的关系及调控机制具有重要临床意义。
NSCLC 的美国国立综合癌症网络(National Comprehensive Cancer Network, NCCN) 指南指出,KRAS突变的NSCLC患者疗效不佳与EGFR-TKI原发耐药相关。目前尚无美国食品药品监督管理局批准的靶向KRAS基因的抗肿瘤药物,阻断KRAS激活后的下游通路是KRAS突变治疗策略的主要探索方向。KRAS下游的信号通路包括PI3K/AKT/mTOR/p70S6K 和RAF/MEK/ERK信号通路。前期研究[17]发现,在NSCLC 中EGFR 可通过PI3K/AKT 上调PD-L1 的表达;最新研究[18]发现,KRAS突变可导致人支气管上皮细胞中MEK/ERK 依赖的PD-L1表达上调,从而介导了肿瘤的免疫逃逸。
mTOR 位于p70S6K 的上游,是信号通路的关键部分。最近研究[19-21]表明mTOR 信号对PD-L1 表达有正向调控作用,而Deng 等[22]研究发现抑制mTOR/p70S6K 信号可提高肿瘤细胞的PD-L1 水平。本研究发现,在KRAS突变的NSCLC 细胞株中p70S6K 被激活,而mTOR、AKT 未被激活。我们推测激活p70S6K 的不是mTOR,可能是上游的一些旁路信号,如磷酸肌醇依赖性蛋白激酶-1(phosphoinositide dependent kinase-1,PDK1) 等。使用p70S6K 抑制剂处理KRAS突变株或WT株,PD-L1的表达较对照组均无明显变化;由此我们推测,在KRAS突变NSCLC细胞株中,p70S6K 的激活与PD-L1的上调没有直接关系。
本研究进一步检测了KRAS突变型NSCLC 细胞株经RAS/RAF/MEK 通路抑制剂处理后PD-L1 蛋白表达的变化。结果发现,在KRAS突变的NSCLC 细胞株中,RAF抑制剂和MEK 抑制剂对PD-L1 的表达存在差异调控。RAF 抑制剂表现为上调PD-L1 的表达,而MEK 抑制剂表现为下调PD-L1 的表达;但由于仅MEK 抑制剂在低浓度时即可同时下调3 个KRASG12V 突变株的PD-L1 表达,且呈一定剂量依赖性,因此我们猜测在KRASG12V 突变株中,KRAS可能通过上调MEK诱导PD-L1的表达。
综上所述,本研究从细胞、组织层面验证了KRASG12V 突变型的NSCLC 患者PD-L1表达较KRAS WT型患者高,并推测KRASG12V 突变的NSCLC 细胞通过上调MEK 信号分子上调PD-L1的表达;因此KRASG12V 突变的NSCLC患者应用PD-1/PD-L1免疫治疗的获益率可能较其他KRAS突变亚型高。本研究为KRAS突变的NSCLC患者治疗策略的制定提供了新的思路。
参·考·文·献
[1] Rothschild SI. Targeted therapies in non-small cell lung cancer: beyond EGFR and ALK[J]. Cancers(Basel),2015,7(2):930-949.
[2] Seigel RL,Miller KD,Jemal A,et al. Cancer statistics,2015[J]. CA Cancer J Clin,2015,65(1):5-29.
[3] Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015[J]. CA Cancer J Clin,2016,66(2):115-132.
[4] Renaud S, Falcoz PE, Schaëffer M, et al. Prognostic value of theKRASG12V mutation in 841 surgically resected Caucasian lung adenocarcinoma cases[J]. Br J Cancer,2015,113(8):1206-1215.
[5] Brahmer J, Reckamp KL, Baas P, et al. Nivolumabversusdocetaxel in advanced squamous-cell non-small-cell lung cancer[J]. N Engl J Med,2015,373(2):123-135.
[6] Borghaei H, Paz-Ares L, Horn L, et al. Nivolumabversusdocetaxel in advanced nonsquamous non-small-cell lung cancer[J]. N Engl J Med, 2015,373(17):1627-1639.
[7] Gettinger S, Rizvi NA, Chow LQ, et al. Nivolumab monotherapy for firstline treatment of advanced non-small-cell lung cancer[J]. J Clin Oncol,2016,34(25):2980-2987.
[8] Horn L,Spigel DR,Vokes EE,et al. Nivolumabversusdocetaxel in previously treated patients with advanced non-small-cell lung cancer:two-year outcomes from two randomized, open-label, phase Ⅲtrials (CheckMate 017 and CheckMate 057)[J]. J Clin Oncol,2017,35(35):3924-3933.
[9] Champiat S, Dercle L,Ammari S, et al. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1[J].Clin Cancer Res,2017,23(8):1920-1928.
[10] Herbst RS, Baas P, Kim DW, et al. Pembrolizumabversusdocetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer(KEYNOTE-010):a randomised controlled trial[J]. Lancet,2016,387(10027):1540-1550.
[11] Fehrenbacher L, Spira A, Ballinger M, et al. Atezolizumabversusdocetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial[J]. Lancet,2016,387(10030):1837-1846.
[12] Kim JH, Kim HS, Kim BJ. Prognostic value ofKRASmutation in advanced non-small-cell lung cancer treated with immune checkpoint inhibitors: a meta-analysis and review[J]. Oncotarget,2017,8(29):48248-48252.
[13] Lan B, Ma C, Zhang C, et al. Association between PD-L1 expression and driver gene status in non-small-cell lung cancer: a meta-analysis[J].Oncotarget,2018,9(7):7684-7699.
[14] Nicholson AG, Chansky K, Crowley J, et al. The International Association for the Study of Lung Cancer Lung Cancer Staging Project:proposals for the revision of the clinical and pathologic staging of small cell lung cancer in the forthcoming eighth edition of the TNM classification for lung cancer[J].J Thorac Oncol,2016,11(3):300-311.
[15] Lococo F, Torricelli F, Rossi G, et al. Inter-relationship between PD-L1 expression and clinic-pathological features and driver gene mutations in pulmonary sarcomatoid carcinomas[J]. Lung Cancer,2017,113:93-101.
[16] Gridelli C, Ardizzoni A, Barberis M, et al. Predictive biomarkers of immunotherapy for non-small cell lung cancer: results from an Experts Panel Meeting of the Italian Association of Thoracic Oncology[J]. Transl Lung Cancer Res,2017,6(3):373-386.
[17] Lastwika KJ, Wilson W, Li QK, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer[J]. Cancer Res,2016,76(2):227-238.
[18] Lee MH, Yanagawa J, Tran L, et al. FRA1 contributes to MEK-ERK pathway-dependent PD-L1 upregulation byKRASmutation in premalignant human bronchial epithelial cells[J]. Am J Transl Res,2020,12(2):409-427.
[19] Cavazzoni A, Digiacomo G, Alfieri R, et al. Pemetrexed enhances membrane PD-L1 expression and potentiates T cell-mediated cytotoxicity by anti-PD-L1 antibody therapy in non-small-cell lung cancer[J]. Cancers(Basel),2020,12(3):666.
[20] Chen MC,Pangilinan CR,Lee CH.Salmonellabreaks tumor immune tolerance by downregulating tumor programmed death-ligand 1 expression[J]. Cancers(Basel),2019,12(1):57.
[21] Chang HL, Kuo YH, Wu LH, et al. The extracts ofAstragalus membranaceusovercome tumor immune tolerance by inhibition of tumor programmed cell death protein ligand-1 expression[J]. Int J Med Sci, 2020,17(7):939-945.
[22] Deng L, Qian G, Zhang S, et al. Inhibition of mTOR complex 1/p70 S6 kinase signaling elevates PD-L1 levels in human cancer cells through enhancing protein stabilization accompanied with enhanced β-TrCP degradation[J]. Oncogene,2019,38:6270-6282.
猜你喜欢
检验医学与临床(2022年19期)2022-10-10
现代临床医学(2022年3期)2022-06-06
健康体检与管理(2022年4期)2022-05-13
天津医科大学学报(2021年4期)2021-08-21
现代临床医学(2021年4期)2021-07-31
昆明医科大学学报(2021年2期)2021-03-29
现代临床医学(2021年2期)2021-03-29
天津医科大学学报(2021年2期)2021-03-29
中国现代医生(2018年22期)2018-12-04
分析化学(2018年12期)2018-01-22
