
特发性肺纤维化中氧化应激调控机制的研究进展
2021-07-02 07:24郭春玉综述张诗晨审校
安徽医科大学学报 2021年6期
郭春玉 综述 张诗晨,方 军,3 审校
特发性肺纤维化 (idiopathic pulmonary fibrosis, IPF) 是一种慢性、进行性、纤维性的间质性肺炎,其病因及发病机制尚未完全阐明。目前其治疗方法多偏重于对症,治疗效果却并不明显,且确诊后五年生存率小于50%,中位生存期仅约3年。IPF的危险因素包括微生物感染、吸烟、放疗或化疗、长期吸入粉尘或石棉等职业和环境暴露等。IPF的病理生理学改变主要是由肺泡上皮细胞反复持续性的微损伤与损伤修复失调,其病理特征主要表现为肺泡上皮细胞损伤、炎性细胞浸润、胞外基质的大量堆积、上皮-间质转化(epithelial-mesenchymal transition, EMT)、成纤维细胞向肌成纤维细胞的转化(fibroblast -myofibroblast transition, FMT),最终造成气体交换及呼吸功能受损。许多研究表明,氧化应激是肺纤维化的主要致病机制之一,IPF的发生与活性氧 (reactive oxygen species, ROS)蓄积导致氧化还原平衡紊乱相关。此外,氧化应激与转化生长因子(transforming growth factor, TGF)-β的交互作用对促进肺纤维化有重要意义。该文主要就氧化应激在IPF的发生发展中相关调控机制的研究进展进行综述。
1 人体内的氧化物及其生物学作用
1.1 氧化物与肺损伤
人体内氧化物与抗氧化物维持动态平衡,当细胞内的抗氧化物不能中和氧化物时发生的不平衡即为“氧化应激”。氧化应激不仅损伤DNA、蛋白质及脂质,还会改变胞内信号通路、甚至基因的表达,此外,还可通过氧化修饰促进异常细胞生长、炎症和其他生理过程。ROS的慢性过量产生可刺激Ⅰ型胶原的过表达,并引发炎症反应。而肺是人体中最容易发生氧化-抗氧化失衡的器官之一,肺的氧分压明显高于心脏、肝脏及大脑中的,而且不断暴露于内源性或外源性氧化物,包括香烟烟雾、空气颗粒物、粉尘、电离辐射等。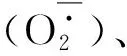
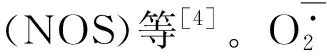
2 氧化应激对炎症的影响
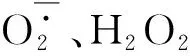
2.2 ROS对巨噬细胞极化的调控作用
此外,氧化应激可诱导巨噬细胞的极化,巨噬细胞的极化在肺组织损伤修复调控中发挥着重要作用。目前的研究认为,在炎症初期巨噬细胞通过分泌促炎因子(如IL-12、IL-1β、IL-2、IL-6、IL-23、TNF-α等)和趋化因子并产生NO和ROS继而经细胞毒作用发挥杀伤病原微生物的作用,这个过程被称为巨噬细胞M1表型(即经典活化型巨噬细胞)极化。随后,巨噬细胞向M2表型(即替代活化型巨噬细胞)极化,M2型巨噬细胞能够促进受损组织的修复与重塑。然而,IPF主要是由持续性的微损伤与损伤修复失调所造成的。在此过程中,M2型巨噬细胞可以:① 促进成纤维细胞的聚集与增殖;② 产生金属蛋白酶组织抑制因子;③ 诱导EMT与FMT,从而促进肺纤维化的进程。超氧化物HO由超氧化物歧化酶(SODs)代谢生成,这个过程可调控巨噬细胞的极化。线粒体氧化应激产生的Cu-SOD、Zn-SOD通过对组蛋白去甲基化酶Jmjd3的氧化还原反应调控来诱导和维持M2型巨噬细胞,从而加速肺纤维化的进程。3 氧化应激对间质纤维化进程调控作用
3.1 ROS对肺纤维化进程的影响
持续性的肺部损伤可产生ROS,而ROS可引起肺泡上皮细胞的凋亡和基底膜的损伤和间质向上皮的转化,破坏肺结构,损害肺泡气体交换等。肺泡Ⅱ型细胞损伤产生ROS引起氧化应激反应,不仅能诱导上皮细胞凋亡,还可以激活细胞内的信号通路,上调促纤维化因子合成与释放,最终导致肺组织损伤和纤维化。同时氧化应激触发的胞内信号能刺激纤维增生和促纤维化因子的表达,针对氧化-抗氧化平衡的干预则可改善肺损伤动物模型的纤维化进展。3.2 NOX4对肺纤维化的调控作用
在肺中与氧化应激调控相关的酶主要是NOX家族。其中,NOX2是呼吸爆发的关键成分,可使巨噬细胞和非特异性免疫系统的其他细胞将氧气转化为ROS用于宿主防御。然而NOX4在体内调控许多生理与病理过程,包括细胞分化、免疫防御与组织纤维化。NOX4被认为是线粒体功能障碍的中介,表明线粒体电子传递链与NOX4两个氧化应激系统之间存在交叉。NOX4介导的氧化应激可调控TGF-β诱导的成纤维细胞的分化、细胞骨架的活力以及转录调节。TGF-β在组织纤维化进程中起着基础性的作用。TGF-β是一种多功能蛋白,其主要生物学作用之一就是促进成纤维细胞的募集与基质蛋白的合成,同时TGF-β还可以通过诱导金属蛋白酶抑制剂的表达抑制基质降解。不仅TGF-β可以诱导NOX4的表达,NOX4依赖的氧化还原反应信号通路同样可以通过前反馈的机制激活TGF-β/Smad信号通路。此外,近年的研究表明NOX4可诱导肺上皮细胞死亡、成纤维细胞分化和胶原沉积,说明NOX4在肺纤维化中发挥着重要作用。同时,NOX4可以介导IPF成纤维细胞对衰老和凋亡的抵抗。有研究表明ROS生成酶NOX4的异常上调,加上核因子E2相关因子2 (nuclear factor elytroid-derived factor 2-related factor, Nrf-2)诱导的缺失,会导致持续的氧化还原失衡,使正常修复的细胞拥有抗凋亡表型。并且在纤维化的衰老小鼠模型中对NOX4的靶向干预能够减弱衰老、抗凋亡的肌成纤维细胞表型,并导致持续性纤维化的逆转。因此,通过靶向干预NOX4的表达可能是治疗或缓解IPF的有效策略。3.3 氧化应激对EMT与FMT进程的影响
EMT与FMT是肺纤维化进展的重要过程。EMT是上皮样细胞向间充质样细胞转化的过程,其主要发生在胚胎形成、组织修复和肿瘤转移过程中。在EMT过程中,上皮细胞标志物如E-钙黏蛋白丢失,同时间充质细胞标志物如整合素、纤维连接蛋白等表达。氧化应激诱导线粒体DNA (mtDNA)损进肺泡上皮细胞凋亡和肺纤维化,其中TGF-β是一种关键且独立的促纤维化细胞因子。在器官纤维化的发展过程中,氧化应激通过激活TGF-β表达参与了对EMT进程的调控。HO是EMT与FMT过程中TGF-β信号通路的中介。有研究表明抑制HIF-1α/TGF-β信号通路对肺泡上皮EMT具有抑制作用。肌成纤维细胞是活化的成纤维细胞,而肌成纤维细胞在表型与结构上与平滑肌细胞相似,具有收缩性、迁移性并表达α-SMA和胞外基质,此外,肌成纤维细胞内氧化产物增多,凋亡敏感性降低。TGF-β通过上调NOX4从而介导HO的产生调控肌成纤维细胞的分化。4 体内的抗氧化应激调控系统
4.1 体内的抗氧化物
人体内所有器官都能表达各种抗氧化物和细胞调控因子,从而参与氧化应激反应。人体内源性的抗氧化防御系统包括:① 低分子质量的抗氧化物,如维生素E、褪黑素、谷胱甘肽(GSH)、尿酸等;② 抗氧化酶,如SODs、过氧化氢酶、谷胱甘肽过氧化物酶(GSH-Px);③ Ⅱ相反应解毒酶,如谷胱甘肽巯基转移酶(GST)同工酶、NADP(H)、醌氧化还原酶(NQO1)等;④ 应激反应蛋白,如血红素加氧酶(HO-1)、铁蛋白等;⑤ 黏蛋白类和金属结合蛋白,如乳铁蛋白、转铁蛋白、金属硫蛋白等;此外,Nrf-2可诱导大多数抗氧化物与解毒酶的表达。这些抗氧化酶以细胞特异性的方式表达于支气管、肺泡上皮细胞和肺巨噬细胞,持续保护肺部免受氧化损伤。4.2 抗氧化反应的关键转录因子Nrf-2
Nrf-2是体内抗氧化反应原件(ARE)的主要调节物,并介导细胞保护性蛋白的表达。体内大量的Ⅱ相反应解毒酶与应激反应蛋白诱导型HO-1都受转录因子Nrf-2的调控,该转录因子是维持细胞氧化还原平衡和减少严重氧化损伤的必要的上游调节因子。Nrf-2属于通过与β-球蛋白基因座控制区高敏感位点NF-E2/AP-1基序结合激活转录调控的蛋白之一,它属于bZip基因的一个子集,共享一个保守的CNC结构域。在哺乳动物中,CNC家族由4个关系密切的蛋白——p45-NFE2、Nrf-1、Nrf-2和Nrf-3,以及Bach1和Bach2两个远亲蛋白组成。Kelch-like ECH相关蛋白1(Keap1)是Nrf-2的天然抑制剂,可将Nrf-2限制在胞质中并通过26 S蛋白酶体促进Nrf-2的降解。应激包括过氧化微环境会损害Keap1对Nrf-2进行泛素化降解的能力,并可诱导新合成的Nrf-2入核与ARE结合。一旦氧化还原反应恢复或有害物质被代谢消除就会引发终止信号,Keap1入核与Nrf-2结合后将其转运回胞质中进行降解。此外,Nrf-2可被磷脂酰肌醇3激酶(PI3K)、PKC、c-Jun、N -末端激酶(JNK)和胞外信号调节蛋白激酶(ERK)磷酸化后上调,而p38 MAPK对Nrf-2通路呈双向调控。Nrf-2参与多种抗氧化相关基因表达的调控,包括:NQO1、HO-1、GSTA1等。Nrf-2可激活对二氧化硅暴露诱导的EMT和TGF-β/Smad信号通路的抑制作用,从而发挥抗氧化与抗纤维化的作用。最近有研究表明,氧化应激、细胞凋亡和炎症都参与早期的肺纤维化进程,而通过激活Keap1/Nrf-2信号通路可起到对肺纤维化保护作用。Nrf-2作为上游转录调控因子对体内抗氧化系统的调控可能是IPF的有效治疗策略。5 结语
目前,IPF的发病机制尚未完全阐明,氧化应激被认为在IPF的进程中扮演着不可或缺的角色。其中,NOX4对肺泡上皮细胞死亡、成纤维细胞分化和胶原沉积的调控在IPF发生发展过程中起着重要作用。相反,Nrf-2-ARE系统所介导的抗氧化相关基因表达是肺免受氧化损伤的重要调控系统之一。因此,针对氧化应激机制相关的治疗策略,如靶向抑制或下调NOX的表达、拮抗NOX4的作用或促进Nrf-2的表达,可能是有效地治疗或缓解IPF的潜在治疗策略。
