
改进的QuEChERS-UPLC-MS/MS法测定动物源性食品中13种全氟化合物
2021-06-22 01:09杜思宇张玉慧王颜红李国琛
食品工业科技 2021年1期
王 莹,杜思宇,张 红,张玉慧,李 玲,王颜红,*,李国琛,*
(1.中国科学院,沈阳应用生态研究所,辽宁沈阳110016;2.农业农村部农产品质量安全环境因子风险评估实验室(北京),北京100029)
全氟化合物(perfluorinated compounds,PFCs)是化合物分子中与碳原子连接的氢原子全部被氟原子取代并带有不同功能团的一类人工合成有机化合物[1]。PFCs主要包括全氟烷基羧酸类(PFCAs)、全氟烷基磺酸类(PFSAs)、全氟烷基磺酰胺类(PFOSAs)、全氟调聚醇(FTOHs)、全氟磷酸及其酯等,其中全氟烷基辛酸(PFOA)和全氟烷基磺酸(PFOS)是两种最受关注的八碳链PFCs[2]。全氟化合物的C-F共价键具有极高的化学键能,不易被水解、光解或生物降解,可在环境中持久性存在、可长距离传输、可沿生物链积累放大[3],是国际上备受关注的新型有机污染物。近年来,在全球生态环境系统[4-6]、生物体[7-8]、人体[9-11]、食品[2,12]等多种介质中均有检出。鉴于PFCs广泛存在性以及具有神经毒性[6,13]、生殖毒性[14]和免疫毒性[15]等毒性效应,国内外研究学者开始关注PFCs对人体的健康风险,并普遍认为膳食暴露是人类摄入PFCs的重要途径[6,16]。人类膳食中水产品、乳及乳制品、肉及肉制品、果蔬等均有不同程度PFCs污染[2,17]。尽管人们已经认识到PFCs可能带来的膳食摄入风险,但国内外关于食品中PFCs的限量标准仍是空白。基于PFCs易与蛋白质结合而累积于生物体组织器官的富集特性[18],膳食占比较大的动物源性食品,特别是肝脏、肾脏、肌肉等蛋白质含量较高的器官组织中富集现象普遍但含量较低(通常在μg/kg级别)[3,19-20]。因此,快速、准确、灵敏的动物源性产品中多种类PFCs含量的检测方法可以为人群暴露于PFCs风险评估提供重要技术支持。
目前,PFCs检测方法主要有气相色谱法(GC)[21]、气相色谱质谱法(GC-MS)[22]、气相色谱-串联质谱法(GC-MS/MS)[23]和液相色谱-串联质谱法(LC-MS/MS)[24-33]。基于PFCs极性大、沸点高、不能直接气化的特点,GC、GC-MS、GC-MS/MS法在分析前需要对PFCs进行衍生化,但处理过程繁琐,易引入污染和干扰物质,这使得上述三种方法在应用上受到局限。HPLC-MS/MS的多反应监测模式(multi-reaction monitoring,MRM),背景干扰少,选择性好、检出限低,在痕量PFCs检测分析中具有显著优势。HPLC-MS/MS对应的前处理方法,主要采用离子对液液萃取、液固萃取、碱消解提取结合弱阴离子交换柱WAX[31]或HLB固相萃取柱净化[29],但是这些传统的前处理方法步骤多、耗时长,重现性差。本实验采用酸化乙腈提取辅以分散固相萃取技术(QuEChERS)进行样品前处理。QuEChERS是目前广泛应用于食品中目标物测定的方法,也已逐步应用到动物源性食品中PFCs检测[24,27-28,30,32],郭萌萌等[27]采用十八烷基键合硅胶(C18)和石墨化碳黑(GCB)分散固相萃取法测定水产品中23种全氟化合物,朱萍萍等[28]、何建丽等[30]、白文荟等[32]采用C18、GCB和N-丙基乙二胺(PSA)3种混合吸附剂净化法测定羊肝、牛肝、猪肝和猪肉中多种全氟化合物。上述改进的QuEChERS-LC-MS/MS方法都具有合理的回收率及较高的灵敏度,但存在样品基质类型不全或检测项目少等缺点。我国人群膳食暴露频繁且PFCs富集普遍的动物源性食品(猪、牛、羊)的肝脏、肾脏、肌肉中PFCAs和PFSAs多种类PFCs同时检测的方法报道较少。同时,国内外颁布的检测方法标准[24-25]也只关注了食品中最典型的PFOA和PFOS,而对同样具有危害性的其它PFCs没有涉及。因此,本实验采用酸化乙腈提取,C18、GCB和PSA混合吸附剂分散固相萃取净化,UPLC-MS/MS检测9种基质(猪、牛、羊的肝脏、肾脏、肌肉)13种PFCs(包括9种PFCAs,4种PFSAs),同位素内标法定量。该方法步骤简化、定量准确、灵敏度高,可广泛应用于动物源性食品中多种PFCs的分析。
1 材料与方法
1.1 材料与仪器
甲醇、乙腈、正己烷 色谱纯,德国Merck公司;甲酸、乙酸铵 色谱纯,美国Fisher Chemical公司;盐酸、氯化钠 优级纯,天津康科德公司;C18、GCB 40~60μm,北京迪马公司;PSA 40~63μm,上海安谱公司;聚丙烯离心管 15、50 mL,美国ThermoFisher Scientific公司;标准品全氟丁烷羧酸(PFBA)、全氟戊烷羧酸(PFPeA)、全氟己烷羧酸(PFHxA)、全氟庚烷羧酸(PFHpA)、全氟辛烷羧酸(PFOA)、全氟壬烷羧酸(PFNA)、全氟癸烷羧酸(PFDA)、全氟十一烷羧酸(PFUd A)、全氟十二烷羧酸(PFDoA)、全氟己烷磺酸钠(PFHxS)、全氟辛烷磺酸钠(PFOS)、全氟癸烷磺酸钠(PFDS)、全氟十二烷磺酸钠(PFDoS)、同位素内标物13C4-PFOA(Perfluoro-n-[1,2,3,4-13C4]octanoic acid,MPFOA)、同位素内标物13C4-PFOS(Sodium perfluoro-1-[1,2,3,4-13C4]octanesulfonate,MPFOS) 均为50μg/mL标准储备液,溶剂甲醇,阴凉避光放置,加拿大Wellington公司;实验所用的动物源性食品 均为市售猪、牛、羊的肝脏、肾脏、肌肉,取可食用部分绞碎并均质,取2.00 g均质样品于50 mL聚丙烯离心管中,待检,其余样品密封保存于-18℃冰箱中。
UltiMate 3000超高效液相色谱仪、TSQ Quantiva三重串联四极杆质谱仪 美国ThermoFisher Scientific公司;T25数显型高速分散机、Vortex genius 3涡流混合器 德国IKA公司;TD25-WS离心机 湖南湘仪公司;THZ-98A恒温振荡器 上海一恒公司;L-119A氮气吹干仪 北京来亨科贸有限公司;Biofuge Stratos台式高速冷冻离心机 美国Thermo Fisher Scientific公司。
1.2 实验方法
1.2.1 标准溶液配制 13种PFCs和2种同位素内标标准中间液的配制:分别准确移取13种PFCs、2种同位素内标标准储备液(50μg/mL)适量于10 mL容量瓶中,甲醇定容,分别配制成浓度均为5μg/mL的标准中间液,浓度以全氟烷基羧酸或全氟烷基磺酸计,-18℃避光保存,有效期12个月。
混合标准工作液的配制:分别准确移取13种PFCs标准中间液各1 mL,于50 mL容量瓶中,甲醇定容。该溶液中每种PFCs浓度为100 ng/mL。-18℃避光保存,有效期3个月。
混合同位素内标中间液的配制:分别准确移取同位素内标中间液MPFOA 1.5 mL、MPFOS 3 mL,于50 mL容量瓶中,甲醇定容。该溶液中MPFOA浓度为150 ng/mL、MPFOS浓度为300 ng/mL。-18℃避光保存,有效期6个月。
混合同位素内标工作液:准确移取混合同位素内标中间液1 mL于10 mL容量瓶中,甲醇定容。该溶液中MPFOA浓度为15 ng/mL、MPFOS浓度为30 ng/mL。-18℃避光保存,有效期3个月。
系列标准工作溶液的配制:准确移取13种PFCs混合标准工作液、混合同位素内标工作液适量,用甲醇配制浓度为0.05、0.1、0.5、1.0、2.0、5.0、10 ng/mL(同位素内标MPFOA浓度为1.5 ng/mL、MPFOS浓度为3.0 ng/mL)的系列标准工作溶液。MPFOA是定量9种PFCAs的同位素内标,MPFOS是定量4种PFSAs的同位素内标。
1.2.2 仪器条件
1.2.2.1 色谱条件 色谱柱:Atlantis T3色谱柱(2.1 mm×150 mm,3μm);流速:0.3 mL/min;柱温:40℃;进样量:10μL;流动相:2.5 mmol/L乙酸铵-甲醇溶液(A)(2.5 mmol/L乙酸铵溶液(B);梯度洗脱程序:0~3 min,90%~40%B;3~12 min,40%~10%B;12~14 min,10%B;14~14.5 min,10%~90%B;14.5~18 min,90%B。
1.2.2.2 质谱条件 电喷雾离子源(ESI);扫描方式:负离子扫描;检测方式:多反应监测(MRM);喷雾电压:3500 V;蒸汽温度;290℃;离子传输管温度:270℃;鞘气流速:30 arb;辅助气流速:10 arb;二级碰撞气:氩气;碰撞气压力:1.5 mTorr;保留时间、定性离子对、定量离子对、碰撞能量见表1。
1.2.3 前处理 称取样品(肝脏、肾脏、肌肉)2 g(精确至0.01 g)于50 mL聚丙烯离心管中,加入混合同位素内标工作液100μL,再加入2 mL水,涡旋混合1 min。加入0.2%盐酸-乙腈溶液10 mL,200 r/min振荡10 min,加入2 g氯化钠,再振荡10 min,5000 r/min离心5 min。将全部上清液转移至15 mL聚丙烯离心管并40℃氮吹至约5 mL,加入100 mg PSA、80 mg C18、30 mg GCB,涡旋混合1 min,振荡10 min,5000 r/min离心10 min。取全部上清液至另一个15 mL聚丙烯离心管中,40℃氮吹至近干,1 mL甲醇定容,15000 r/min离心5 min,上清液供上机测定。
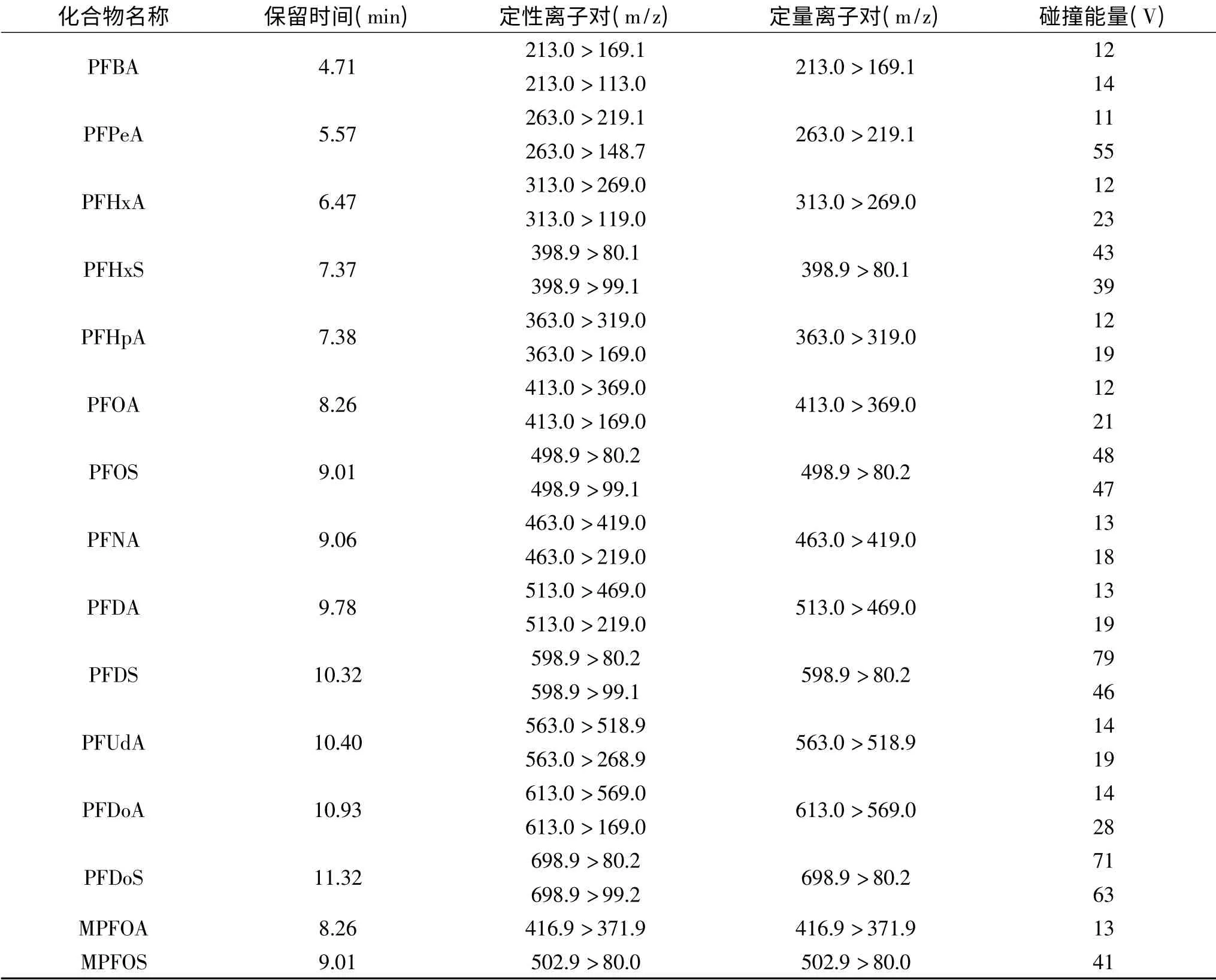
表1 13种全氟化合物的质谱参数Table 1 Mass spectrometric conditions of 13 PFCs
为减少实验过程中带来的污染及背景干扰,全程尽量避免使用聚四氟乙烯器皿而使用聚丙烯材质器皿[28]。
1.3 方法学考察
1.3.1 基质效应 本实验采用1.2.1配制的系列标准工作溶液,按照1.2.2条件测定,绘制标准曲线。同时,选取9种空白样品基质(猪肉、猪肝、猪肾、牛肉、牛肝、牛肾、羊肉、羊肝、羊肾)不加同位素内标按照1.2.3前处理后,采用空白基质溶液作为溶剂配制相同浓度范围的系列基质标准工作溶液,按照1.2.2条件测定,绘制标准曲线。通过比较基质标准曲线与溶剂标准曲线方程斜率的比值来判断基质效应的影响。
1.3.2 线性范围、检出限、定量限 0.05、0.1、0.5、1、2、5、10 ng/mL系列标准工作溶液按照1.2.2条件测定,同位素内标法定量,以各个目标物与对应的同位素内标物的峰面积比值为纵坐标、以各目标物浓度与与对应的同位素内标物的浓度比值为横坐标,求线性方程及相关系数。按照GB/T 27417-2017《合格评定 化学分析方法确认和验证指南》[34]中5.4条款规定,在猪、牛、羊的肝脏、肾脏、肌肉空白样品基质中添加预估最低可接受浓度0.15μg/kg,每个样品平行测定10次,计算方法检出限(LOD)=0+3SD、方法定量限(LOQ)=0+10SD。
1.3.3 回收率和精密度 按照GB/T 27404-2008《实验室质量控制规范 食品理化检验要求》[35],对于未制定MRL限量的物质,回收率实验应在方法测定低限、常见限量指标、一个合理的添加浓度三个水平进行。本实验采用在猪肝、牛肝、羊肝、猪肾、牛肾、羊肾、猪肉、牛肉、羊肉9个不同基质空白样品中添加0.2、1和2μg/kg三个浓度,每个浓度6个平行,前处理方法参照1.2.3,仪器条件参照1.2.2,同时做空白实验,均扣除本底值后计算回收率及精密度。
1.4 数据处理
采用美国ThermoFisher Scientific公司Xcalibur 3.0软件、Origin 8.0版本进行数据处理。
2 结果与分析
2.1 色谱和质谱条件优化
2.1.1 色谱条件优化 PFCs具有亲水性和疏水性以及一定的表面活性和极性,相关标准[24-26]及文献[27-30]多数采用较低硅羟基活性填料的C18液相色谱柱分离、乙腈-乙酸铵水溶液或甲醇-乙酸铵水溶液流动相梯度洗脱,来实现这类化合物的良好分离。本实验重点考察两种流动相体系乙腈-乙酸铵水溶液、甲醇-乙酸铵水溶液对PFCs及同位素内标的灵敏度差异。通过比较相同浓度标液峰面积(见图1)发现,两种流动相梯度洗脱条件下,15种物质峰形均尖锐对称。与乙腈-乙酸铵水溶液流动相相比,以甲醇-乙酸铵水溶液为流动相时,除PFBA峰面积减少29%之外,其他14种物质的色谱峰峰面积增加了22%~151%。所以,本实验选择峰面积较大、灵敏度较高的甲醇-乙酸铵水溶液作为流动相体系。

图1 甲醇-乙酸铵水溶液、乙腈-乙酸铵水溶液流动相条件下的标准溶液(10 ng/mL)峰面积比较Fig.1 Comparison of peak area of standard solution(10 ng/mL)in methanol-ammonium acetate and acetonitrile-ammonium acetate mobile phase system
13种PFCs及2种同位素内标物是羧酸或磺酸盐,在流动相中加入乙酸铵,能使溶液保持一定的pH及离子强度,可以有效地增强电喷雾离子化响应值,并减少溶剂效应,改善峰形[32];但也有研究表明,较高浓度的乙酸铵对质谱检测有较强的抑制作用[36]。标准[24-26]及文献[27-30]中常见的乙酸铵浓度为1~10 mmol/L,本实验以甲醇和水为溶剂,分别配制相同浓度的乙酸铵甲醇溶液及乙酸铵水溶液。通过比较1、2.5、5、10 mmol/L四个不同浓度条件下的测定结果(见图2)可以看出,PFOS和PFDoA在1、2.5 mmol/L浓度的峰面积较高且几乎相等,其余13种物质均在乙酸铵浓度为2.5 mmol/L时,色谱峰面积达到最大值。
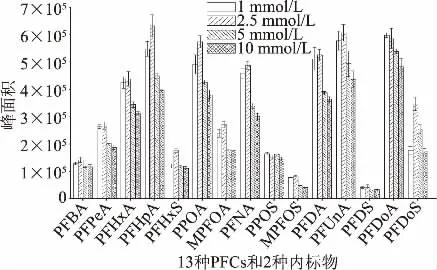
图2 不同浓度乙酸铵对标准溶液(5.0 ng/mL)峰面积的影响Fig.2 Effect of different concentrations of ammonium acetate on peak area of standard solution(5.0 ng/mL)
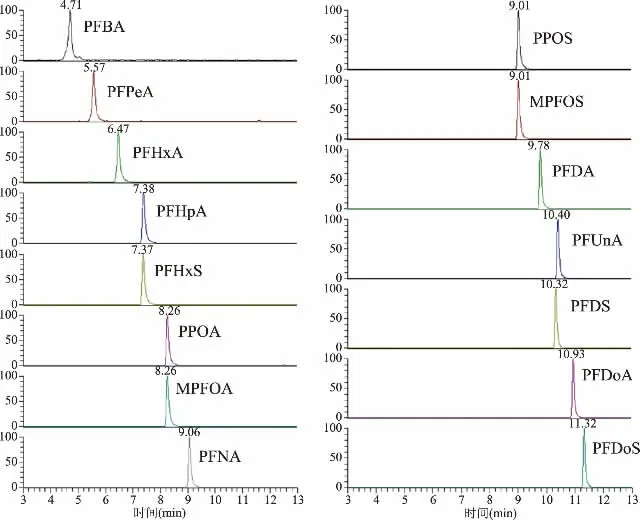
图3 13种PFCs和2种同位素内标标准溶液(1.0 ng/mL)MRM色谱图Fig.3 MRM chromatograms of 13 PFCs and 2 internal standards(1.0 ng/mL)
因此,本实验采用Atlantis T3色谱柱,以2.5 mmol/L乙酸铵甲醇溶液-2.5 mmol/L乙酸铵水溶液进行梯度洗脱。由图3可见15种物质在12 min内全部流出,目标化合物保留时间适中,峰面积、信噪比较高,杂峰响应较小;在空白基质中添加目标化合物,在上述检测条件下,谱图无干扰杂峰,13种PFCs响应好。随碳链的增加,PFCs在C18柱上的保留逐渐增强,同一碳链数全氟磺酸类化合物保留强于全氟羧酸类化合物。PFBA最先流出,PFDoS最后流出。
2.1.2 质谱条件选择 PFCs化学结构中具有羧基或磺酸基,因此采用负离子模式检测。本实验采用流动注射进样方式,分别对5μg/mL标准溶液进行m/z 200~1000 ESI一级全扫描,由实验结果发现,PFCs主要以电离后失去羟基上氢原子[M-H]-最强,确定其为准分子离子,并优化喷雾电压、蒸汽温度、离子传输管温度以获得较强的响应值。接下来,以其作为母离子进行子离子扫描并优化碰撞能量,确定定性离子和定量离子。在实验过程中,PFCAs生成[M-H-44]-子离子,推断是PFCAs发生中性丢失CO2产生;PFSAs生成m/z 99和m/z 80子离子,推断是PFSAs断裂生成[FSO3]-和[SO3]-。本次实验选取丰度较高且干扰较少的子离子[M-H-44]-和[SO3]-分别作为PFCAs和PFSAs的定量离子。以MRM模式采集数据,具体参数见表1。
2.2 前处理优化
针对动物源性食品(肝脏、肾脏、肌肉)基质复杂、含有大量蛋白质及内源性物质等特点,本研究采用改良的QuEChERS样品前处理方法,酸化乙腈提取,C18、PSA和GCB混合吸附剂分散固相萃取净化,减少基质中杂质干扰、提高回收率。
2.2.1 提取溶剂优化 已报道的文献中采用乙腈[29]、盐酸-乙腈[28,31]作为提取溶剂提取动物源性食品的肌肉和肝脏基质中的PFCs。鉴于乙腈是常用的动物源性食品有机提取溶剂,而且PFCs是酸性化合物,在酸性环境下呈非解离状态,有利于进入有机相,所以本实验选择盐酸-乙腈作为提取溶剂。
同时,考察了0.05%、0.1%、0.2%、0.3%四个不同浓度盐酸-乙腈对动物源性食品中13种PFCs的提取回收率。由图4结果表明,在0.05%~0.3%范围内,9种PFCAs的回收率基本是逐渐增加趋势,4种PFSAs是逐渐增加再减少趋势,0.1%、0.2%、0.3%盐酸-乙腈的回收率均在合理范围内,分别为76.3%~114.6%、95.3%~119.4%、85.8%~118.9%。上述三个浓度都是适宜的提取浓度,但0.2%盐酸-乙腈的回收率较集中,分布在95%~120%之间,最低回收率95.3%均高于其他两个浓度;同时为避免增加盐酸用量对后续分析产生干扰,所以选择0.2%盐酸-乙腈作为提取溶剂。在提取过程中,0.2%盐酸-乙腈使样品基质中大量蛋白质变性形成沉淀,并通过高速离心去除。但含脂量较高的样品进行蛋白沉淀时,易将样品中的脂肪和水溶性杂质也提取出来,造成提取液混浊,可能对LC-MS/MS分析产生基质干扰,因此对提取液要进一步净化。
2.2.2 净化方式优化 QuEChERS方法是使用分散固相萃取技术净化,通过将固相吸附剂直接加入到样品提取液中来达到吸附干扰物质的目的。动物源性食品中存在蛋白质、碳水化合物、色素、脂肪、甾醇等物质,可以通过采用不同类型吸附剂达到净化效果。已有文献[28,32]选用C18、PSA、GCB三种吸附剂单一或不同配比进行PFCs净化,但存在样品基质种类较少或检测项目少等缺点。本实验比较了3种吸附剂单一和混合方式对动物源性食品的肝脏、肾脏和肌肉中多种PFCs的净化效果及回收率。
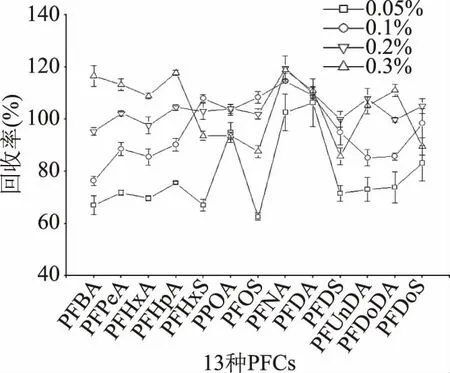
图4 不同浓度盐酸-乙腈提取溶剂对13种PFCs提取效率Fig.4 Extraction efficiencies of 13 PFCs by different concentions of HCl-acetonitrile
2.2.2.1 C18优化 C18主要吸附脂肪和酯类等非极性共萃物[37]。通过在空白样品中添加目标物,做2μg/kg添加浓度3个平行,比较40~100 mg单一吸附剂C18对PFCs回收率的影响,发现随着C18用量由40 mg增加到80 mg,回收率亦增加,在80 mg达到88.4%~116.3%(见图5);由80 mg增加到100 mg,大多数PFCAs回收率基本保持不变,4种PFSAs和PFHpA、PFDA回收率有所下降,但13种PFCs回收率仍处于85.5%~118.0%的合理范围(见图5)。因此采用合理回收率的最小用量80 mg。但净化后的溶液有色素残留,会污染色谱柱及检测仪器,需要进一步去除色素。
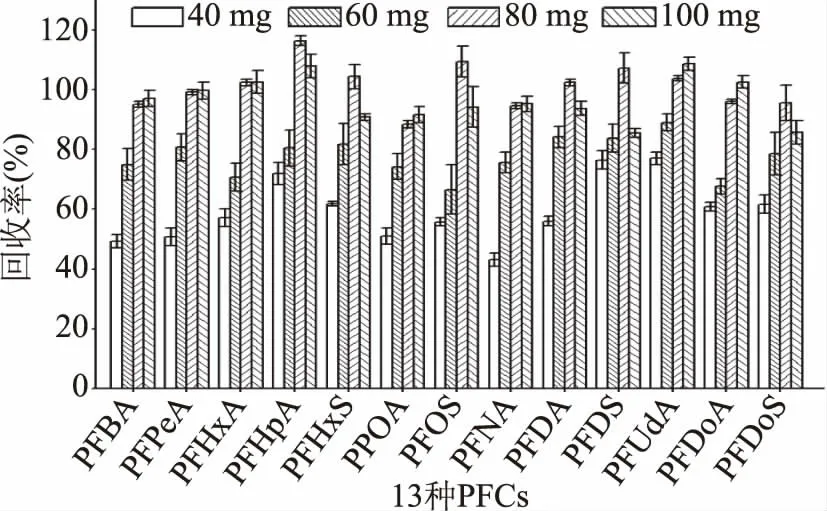
图5 C18用量对PFCs回收率的影响Fig.5 Effect of C18 amount on recoveries of PFCs
2.2.2.2 PSA优化 PSA是一种弱阴离子交换剂,可以吸附碳水化合物、有机酸、少量色素等极性干扰物质[37]。在空白样品中添加目标物,做2μg/kg添加浓度3个平行,在80 mg C18净化基础上,同时加入80~140 mg PSA观察PFCs的回收率变化情况。由图6可以得知随着PSA用量的增加,PFCAs回收率呈先上升后下降趋势,并在100、120 mg时回收率达到合理最大值;PFSAs回收率呈下降趋势,80、100 mg回收率是83.9%~92.1%。综合考虑,选择100 mg PSA为最佳用量。但净化后的溶液有色素残留,需要加入GCB去除。
2.2.2.3 GCB优化 GCB主要吸附色素、甾醇等杂质,通过π键作用来吸附与分离不同化合物。PFCs的π电子被高电负性的氟束缚,不能与GCB形成π键而被解析;而不含氟的化合物则可与GCB形成π键而吸附,从而达到吸附杂质净化样品的目的[38]。
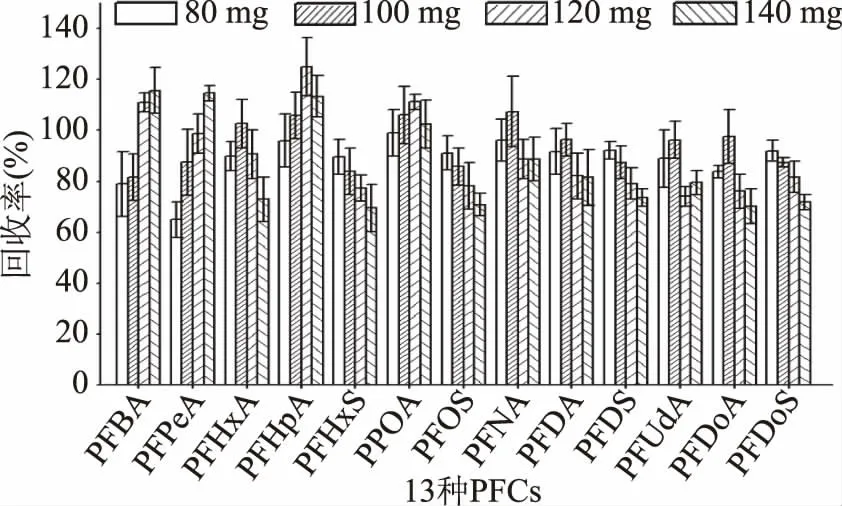
图6 PSA用量对回收率的影响Fig.6 Effect of PSA amount on recoveries of PFCs
通过在空白样品中添加目标物,做2μg/kg添加浓度3个平行,考察不同GCB用量与80 mg C18、100 mg PSA组合对13种PFCs加标回收率的影响,结果如图7所示。随着GCB用量增加,回收率最大值基本保持不变,在110.2%~121.6%之间;最小值变化较大,为53.6%~84.3%。GCB用量为30、40、50 mg时,回收率为75.6%~114.0%,均在合理范围内;其中30 mg的回收率在84.3%~110.2%之间,不同PFCs的回收率变化幅度不大。鉴于上述3个添加量都可以有效去除色素、样品溶液接近无色的前提下,考虑到增加GCB吸附剂用量可能会带来实验成本增加及杂质引入,最后确定30 mg是最优化的GCB添加量。
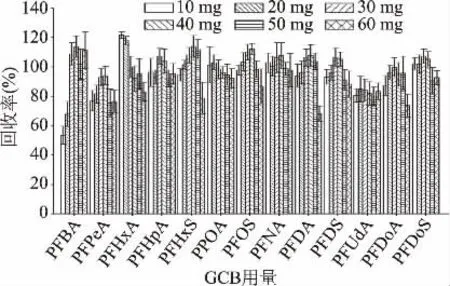
图7 GCB用量对PFCs回收率的影响Fig.7 Effect of GCB amount on recoveries of PFCs
2.3 方法学考察
2.3.1 基质效应 液质检测过程,样品基质本身的内源性成分及前处理过程中引入的外源性成分会改变待测物的离子化效率,进而影响检测方法的灵敏度和选择性,即存在“基质效应”(matrix effect,ME)。目前常用的基质效应评价方法是采用基质标准曲线的线性方程斜率与溶剂标准曲线的线性方程斜率的比值,可用百分数表示。当ME为80%~120%,说明存在基质效应,但影响不大;当50%<ME<80%或120%<ME<150%时,表现为中等程度的基质效应;当ME<50%或ME>150%,表示基质效应强烈[39]。
由实验结果可知,9种基质中13种PFCs基质效应均不相同,变化范围是81%~119%;以PFOA为例(见表2),ME为86%~111%。上述结果表明待测目标物在9种基质中存在一定的基质效应,但影响不明显。因此,本方法采用溶剂甲醇配制系列标准工作溶液进行定量。
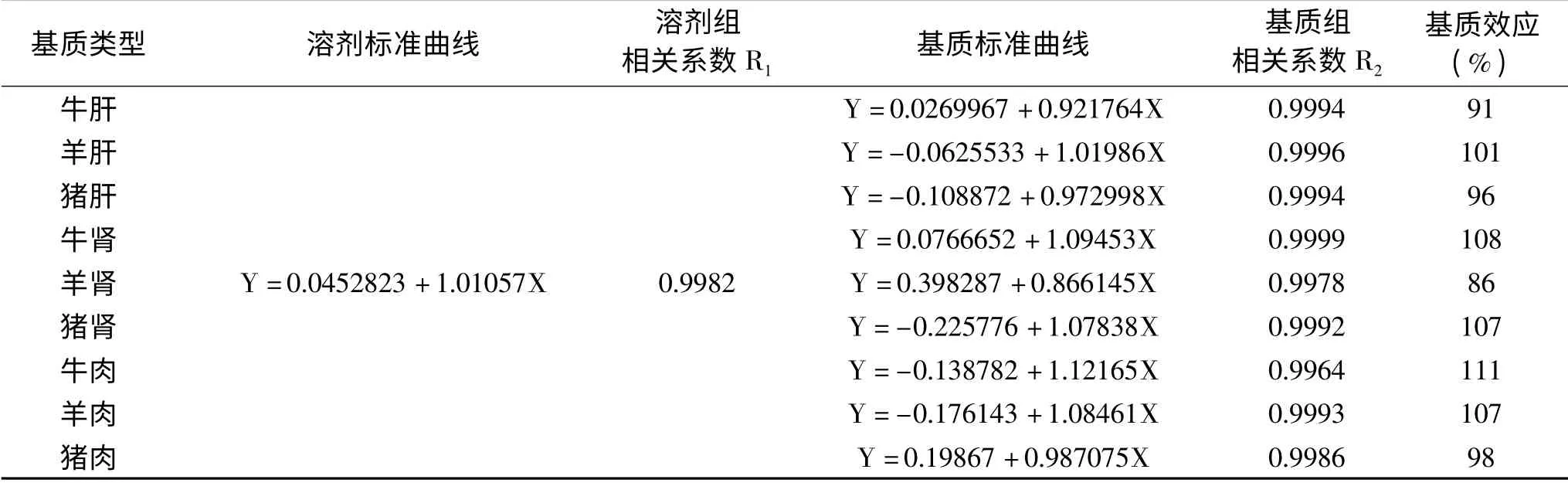
表2 PFOA基质效应Table 2 Matrix effects of PFOA
2.3.2 线性范围、检出限、定量限 实验结果表明,13种PFCs在0.05~10 ng/mL浓度范围内线性关系良好,相关系数为0.9974~0.9999,均大于0.99。在空白样品中添加预估最低可接受浓度0.15μg/kg,每个样品平行测定10次,分别计算3倍标准偏差、10倍标准偏差,确定本方法LOD和LOQ分别为0.02~0.05、0.06~0.15μg/kg。具体见表3。
2.3.3 回收率和精密度 13种PFCs在0.2、1、2μg/kg三个添加水平的平均回收率为62.3%~119.3%,相对标准偏差(RSD)分别为6.4%~19.9%、5.7%~18.3%、3.5%~15.0%(见表4~表6)。本方法具有较高的回收率和精密度,满足GB/T 27417-2017[34]中多残留分析要求。
2.4 实际样品的测定结果
采用优化的前处理方法和检测条件,对市场上购买的16个样品进行13种PFCs分析测定。实验结果表明(见图8),13种PFCs的含量为0.046~5.1μg/kg;PFHxA和PFOS分别有9个样品检出,检出率高达50%;PFBA、PFHp A、PFOA、PFNA、PFDS的检出率均大于20%。由此可见,PFCs在动物源性食品中,尤其是肝脏、肾脏、肌肉中是普遍存在的,这与文献报道基本一致[2,19-20]。
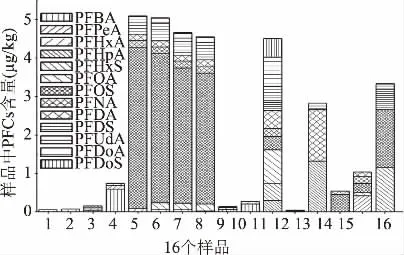
图8 16个动物源性食品中13种全氟化合物的含量Fig.8 Contents of 13 PFCs in 16 animal-derived foods
3 结论
本文建立了UPLC-MS/MS同时测定动物源性食品(猪、牛、羊的肝脏、肾脏、肌肉)中13种全氟化合物(包括9种PFCAs,4种PFSAs)的检测方法。样品采用0.2%盐酸-乙腈提取,C18、PSA、GCB混合吸附剂的分散固相萃取技术净化,同位素内标法定量。结果显示,13种PFCs在12 min内得到有效分离,相关系数均大于0.99;本方法具有较高的灵敏度和精密度,检出限为0.02~0.05μg/kg,定量限为0.06~0.15μg/kg,0.2、1、2μg/kg三个添加浓度平均回收率为62.3%~119.3%,相对标准偏差为3.5%~19.9%。本方法简单、实用性强、分析速度快,可广泛应用于动物源性食品中PFCs的分析,同时为PFCs的风险评估提供重要的技术支持。
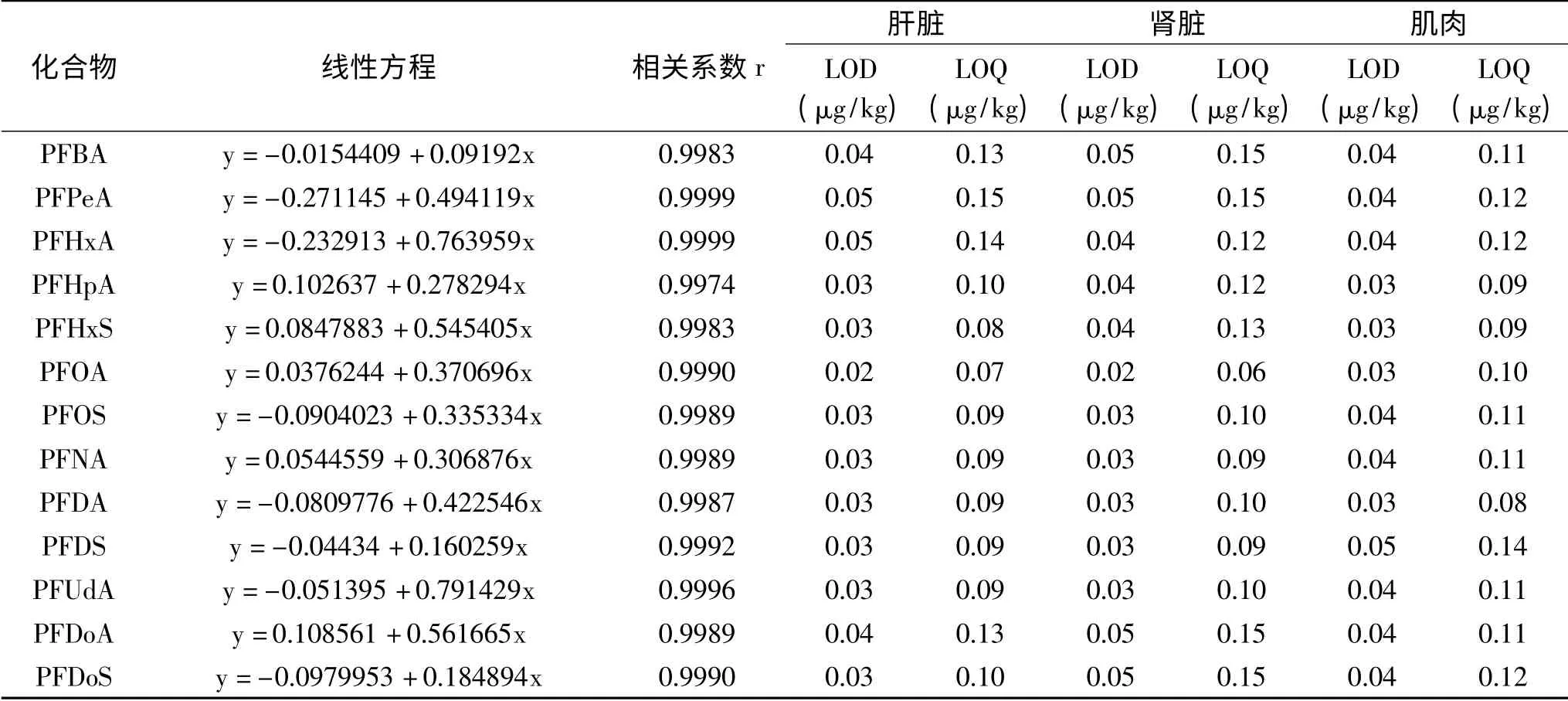
表3 13种PFCs线性方程、相关系数、检出限和定量限Table 3 Linear equations,correlation coefficients,LODs and LOQs for 13 PFCs
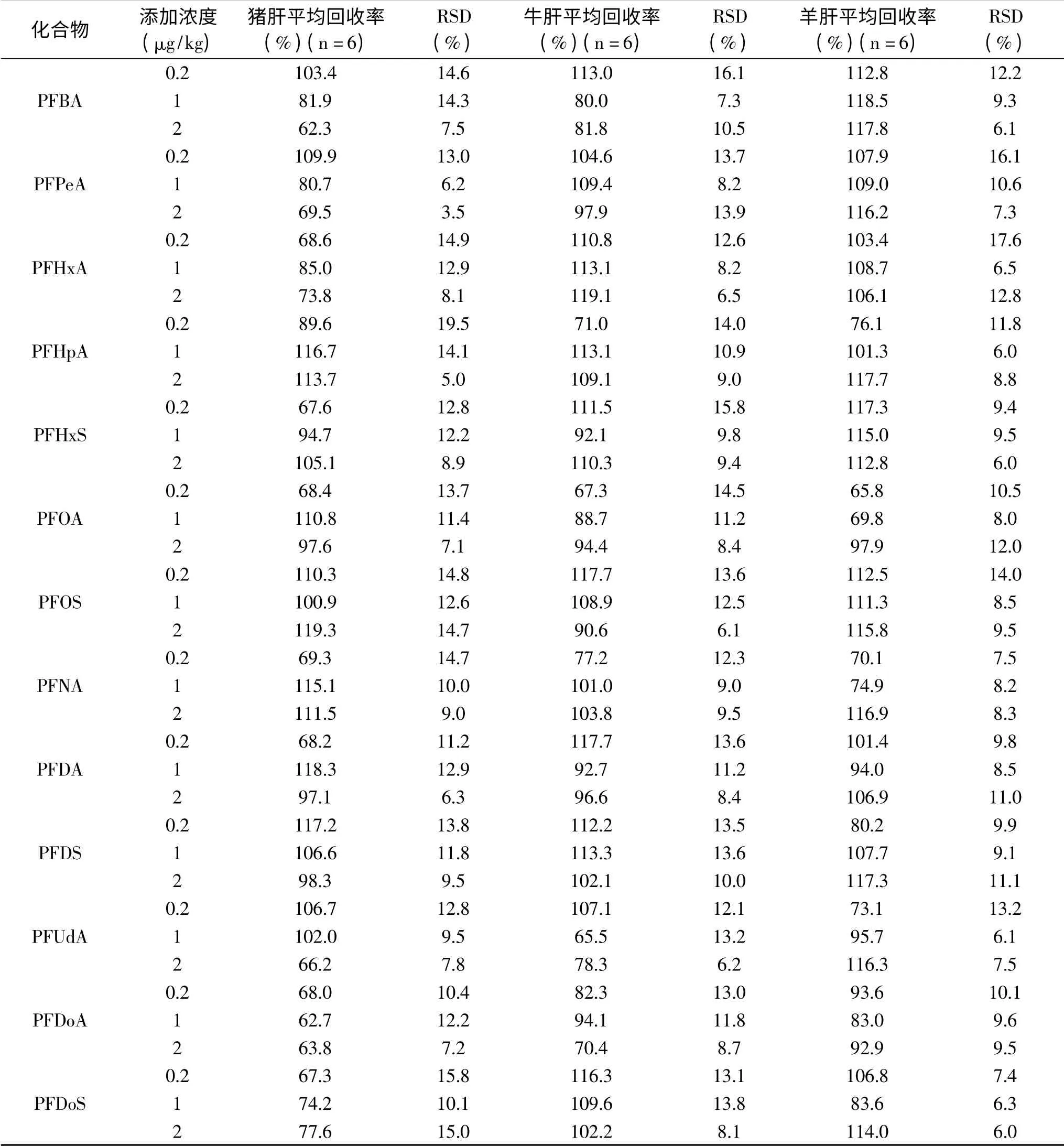
表4 猪肝、牛肝、羊肝中13种PFCs的平均回收率和相对标准偏差Table 4 Mean recoveries and relative standard deviations of 13 PFCs in pig liver,cattle liver and lamb liver
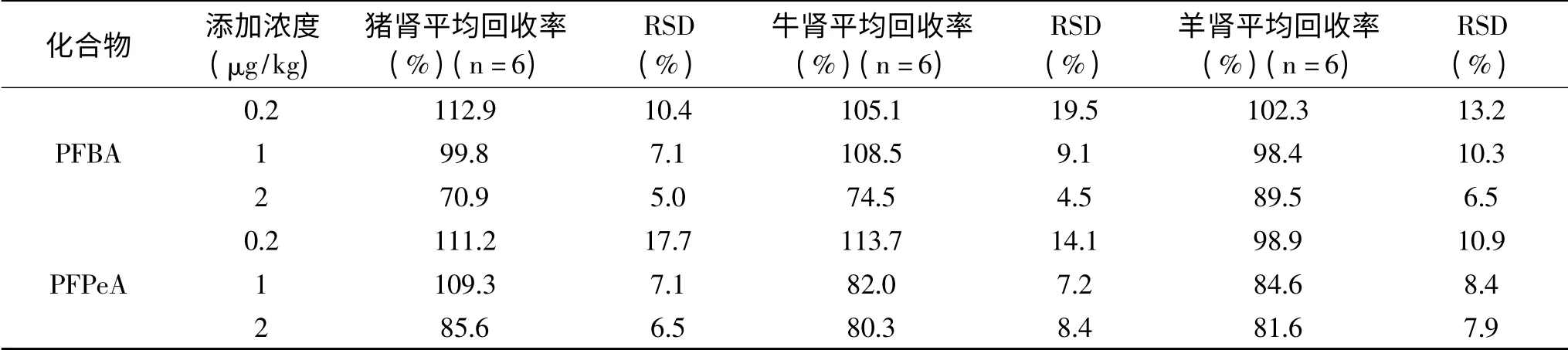
表5 猪肾、牛肾、羊肾中13种PFCs的平均回收率和相对标准偏差Table 5 Mean recoveries and relative standard deviations of 13 PFCs in pig kidney,cattle kidney and lamb kidney
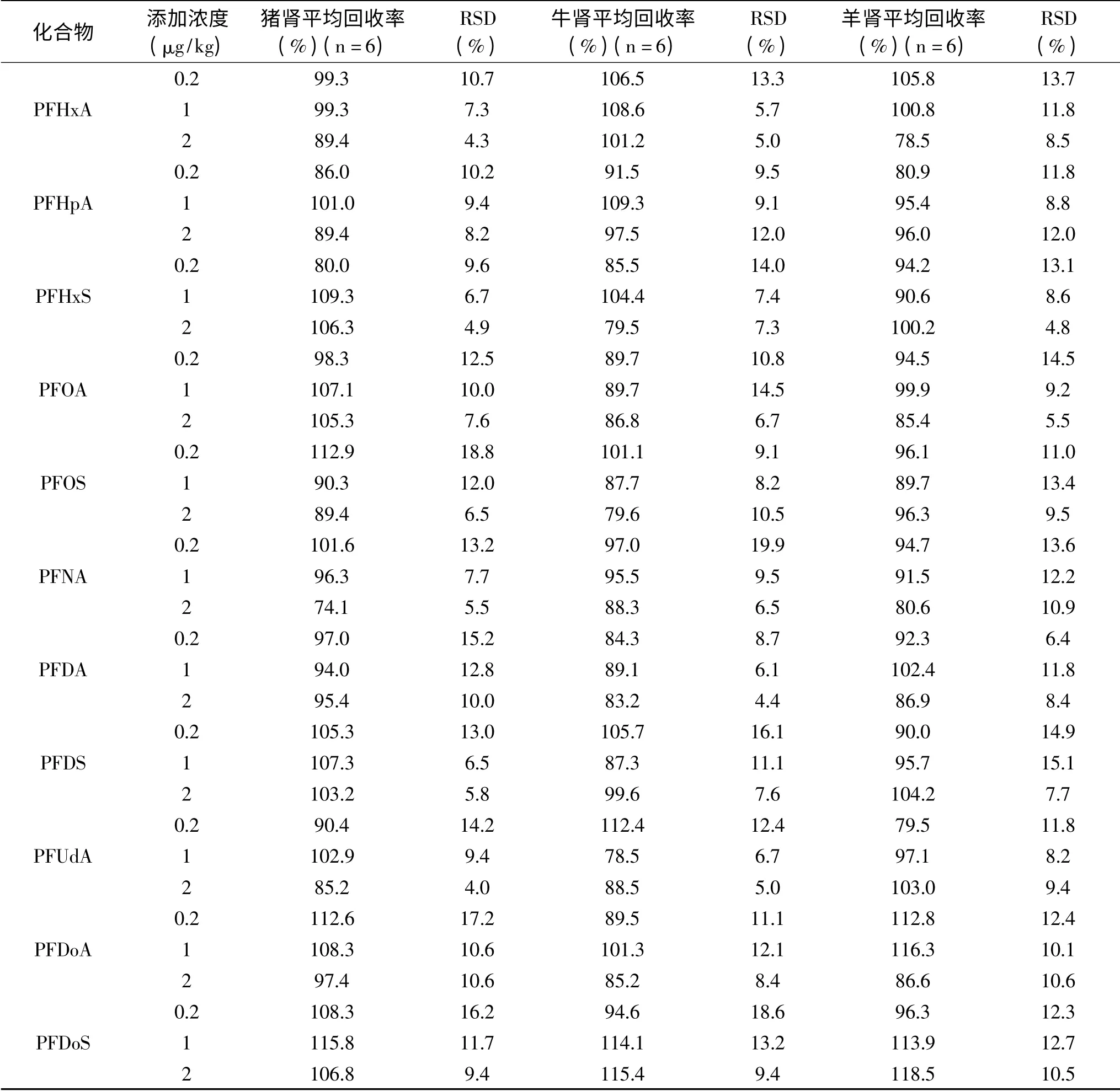
续表
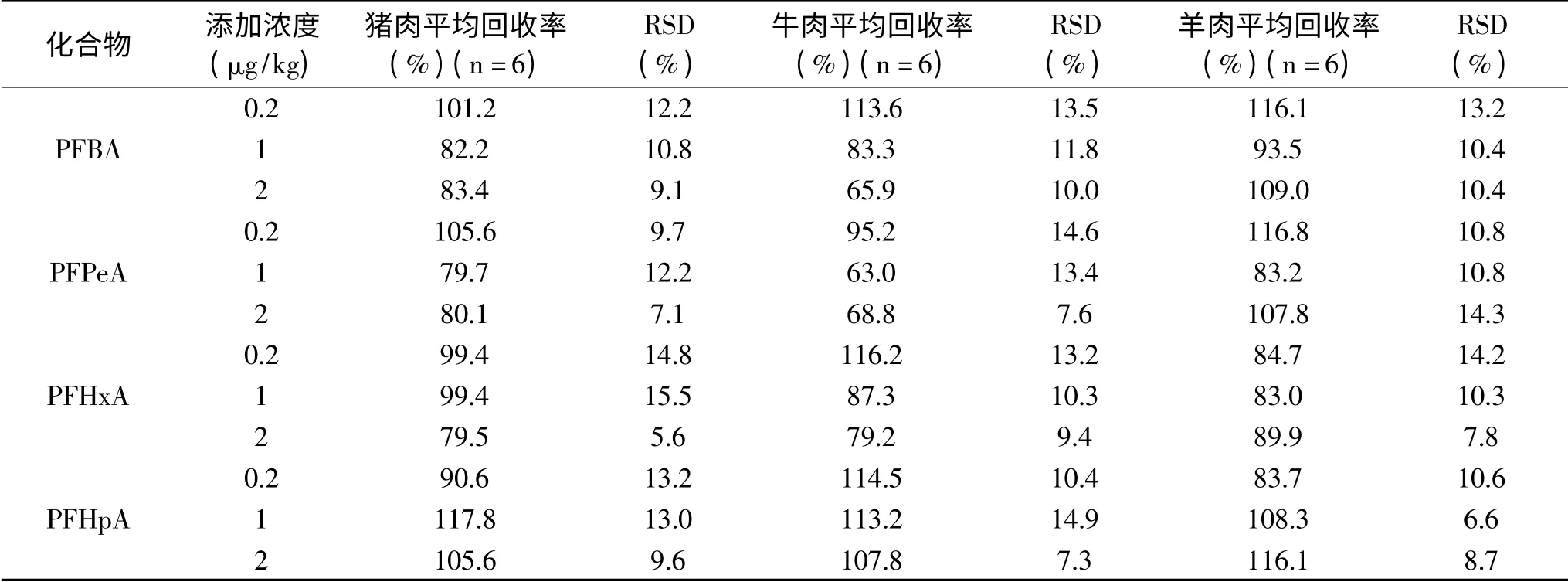
表6 猪肉、牛肉、羊肉中13种PFCs的平均回收率和相对标准偏差Table 6 Mean recoveries and relative standard deviations of 13 PFCs in pork,beef and mutton
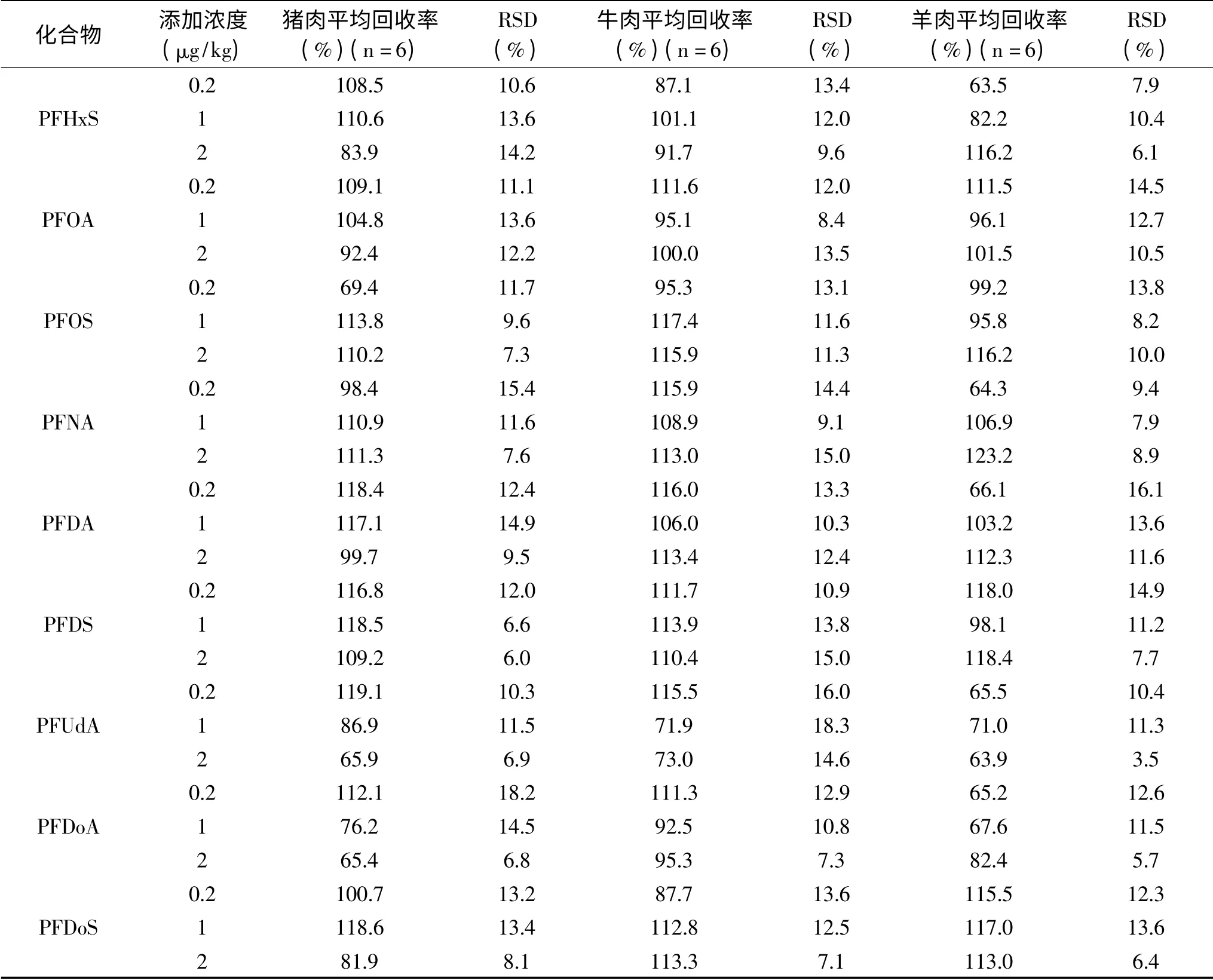
续表
猜你喜欢
现代仪器与医疗(2022年2期)2022-08-11
湖南农业大学学报(自然科学版)(2020年5期)2020-11-11
食品安全导刊(2020年21期)2020-09-07
化工学报(2020年7期)2020-07-21
农家科技中旬版(2019年9期)2019-10-08
中国中医急症(2019年10期)2019-05-21
分析化学(2019年3期)2019-03-30
造纸化学品(2017年1期)2017-01-21
山东工业技术(2016年13期)2016-06-29
分析化学(2015年7期)2015-07-30
