
高效液相色谱法测定北沙参中的蔗糖及其指纹图谱
2021-06-22 08:29苏本正都波周建永
化学分析计量 2021年6期
苏本正,都波,周建永
(1.山东省中医药研究院,济南 250014; 2.天俱时工程科技集团有限公司山东分公司,济南 250101)
高效液相色谱以其灵敏、快捷、可分离大多数液体样品等优点,广泛应用于中药有效成分含量测定、组分分析、指纹图谱等方面[1]。紫外检测器(UV)具有高灵敏度和稳定性,因此高效液相色谱中应用较为广泛,但是对于没有紫外吸收或紫外末端吸收的有机化合物,其无法检测。蒸发光散射检测器(ELSD)弥补了上述紫外检测器的不足,显示出极大的优越性[2–4]。2020年版《中国药典》已经在黄芪、四季青、酸枣仁、山银花等20种中药材的含量测定项目中使用蒸发光散射检测器[5]。
北沙参是伞形科植物珊瑚菜(Glehnia littoralis Fr.Schmidt ex Miq.)的干燥根,为常用中药,可缓解口燥咽干、肺热咳嗽等病症[6–8],其主要化学成分为糖类、香豆素类、磷脂、生物碱、氨基酸、挥发油、微量元素等[9–11]。有研究表明,北沙参中的糖类成分含量最高[12],具有较好的抗肿瘤、抗氧化以及免疫调节活性[13–15]。因此对北沙参中糖类成分进行测定,可以有效评价北沙参的质量,保证临床用药有效性。中药指纹图谱是一种综合的、可量化的鉴定手段,可从整体上鉴别评价中药材及中药制剂半成品质量的真实性、优良性、稳定性[16–17]。成分测定与指纹图谱联合使用,可以全面评价中药的质量。
目前法定标准中没有北沙参的含量测定和指纹图谱检测项目,为了增强标准的可控性,笔者建立了北沙参的指纹图谱测定方法和蔗糖的含量测定方法,以期为制订北沙参质量标准奠定基础。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:Waters 2695型,附带Waters 2420型蒸发光散射检测器和Empower 色谱工作站,美国waters公司。
精密电子天平:AGBP210S型,感量为0.000 01 mg,德国Sartorius公司。
乙腈:色谱纯,美国赛默飞世尔公司。
实验所用其余试剂均为分析纯。
实验用水为超纯水。
蔗糖对照品:50 mg,编号为1507–200001,中国药品生物制品检定研究院。
北沙参药材编号和来源见表1。

表1 北沙参药材样品来源表
1.2 对照品溶液的制备
取蔗糖对照品10 mg,精密称定,加水定容至10 mL的量瓶中,配制成1 mg/mL的蔗糖对照品溶液,摇匀,用0.45 µm微孔滤膜滤过,即得。
1.3 样品溶液的制备
取北沙参样品粉末(过0.42 mm筛)4 g,精密称定,加40%乙醇240 mL,超声提取(功率250 W,频率53 kHz)30 min,滤过,取续滤液100 mL,浓缩至10 mL,0.45 µm微孔滤膜滤过,即得。
1.4 色谱条件
色 谱 柱:Innoval NH2色 谱 柱(250 mm×4.6 mm,5 µm,北京艾杰尔科技有限公司);流动相:乙腈–水(80∶20),等度洗脱;流量:1 mL/min;柱温:30℃;ELSD的漂移管温度:60℃;喷雾器加热级别:100%;气体压力:137.90 kPa。
2 结果与讨论
2.1 蒸发光散射检测器系统条件的优选
2.1.1 正交试验因素、水平设计
蒸发光散射检测器的系统条件由喷雾器加热级别、漂移管温度、气体压力和增益4个条件组成,由于增益值仅影响信号放大的程度,对检测结果没有影响,所以本试验以漂移管温度A、喷雾器加热级别B、气体压力C为3个考察因素,每个因素分别设有三个水平,通过正交试验对检测器条件进行优选,因素水平见表2。

表2 因素水平表
2.1.2 正交试验方法与结果
精密吸取对照品溶液10 µL,注入高效液相色谱仪进行检测。以蔗糖的峰面积作为评价指标,优选条件。结果见表3~表5。

表3 正交试验安排表

表4 正交试验结果表

表5 方差分析表
由表5可知,A、C两个因素P值均小于0.05,说明漂移管温度、气体压力这两个因素对蔗糖的测定均具有显著性意义;B因素的P值大于0.05,说明喷雾器加热级别对蔗糖的测定不具有显著性意义。根据试验结果进行直观分析,A1B3C1为最佳选择,即漂移管温度为60℃,喷雾器加热级别为100%,气体压力为137.90 kPa。
2.2 色谱条件选择与色谱图
氨基柱其固定相是以超纯全多孔球形硅胶为基质,通过采用独有的固定相键合技术,使用含有氨基丙基官能团的有机硅烷键合而成,能比硅胶柱更快达到平衡,对流动相的水含量没有硅胶柱敏感。氨基柱可用于极性化合物的正相分析,弱阴离子交换以及含水的反相液相分析。氨基柱柱被广泛应用于木糖、乳糖、葡萄糖等糖类的反相分析模式中。故本实验选用氨基柱作为色谱柱。
分别吸取蔗糖对照品溶液和样品溶液各10µL,注入高效液相色谱仪检测,开启ELSD采集信号,得到蔗糖对照品和样品的蒸发光检测色谱图,见图1、图2。由图1、图2可知,对照品图谱和样品图谱中,ELSD检测到蔗糖峰在11.5 min附近。
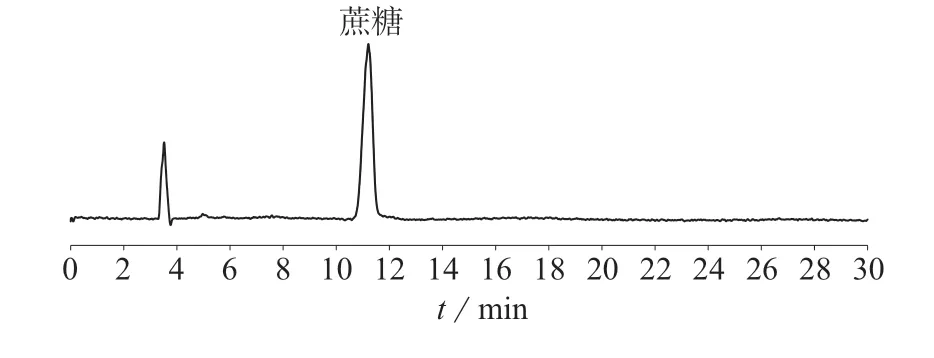
图1 蔗糖对照品的HPLC–ELSD图
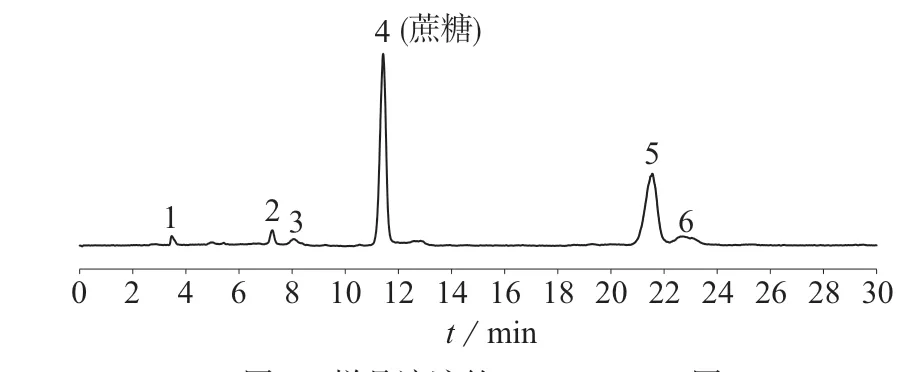
图2 样品溶液的HPLC–ELSD 图
2.3 线性关系与检出限
取蔗糖对照品溶液,分别进样2.5、5、10、15、20、25 µL,经ELSD检测,以蔗糖进样质量的自然对数值为横坐标X,以蔗糖峰面积的自然对数值为纵坐标Y,进行线性回归计算得线性回归方程为Y=1.118 0X+11.86(r=0.999 7),结果表明,蔗糖进样质量在7.30~72.95 µg范围内线性关系良好。将蔗糖对照品溶液逐级稀释,分别进样分析,以3倍信噪比计算检出限,计算得检出限为0.02 mg/mL。

表6 蔗糖经ELSD检测的线性关系数据
2.4 精密度试验
取S2号样品溶液,连续进样6次,进行HPLC指纹图谱分析。分别记录各共有峰的保留时间和峰面积,并对其峰面积结果进行统计分析,结果见表7(其中4号峰为蔗糖色谱峰)。表7结果表明,各共有峰峰面积的相对标准偏差均小于3%,说明该方法精密度良好。

表7 精密度试验结果
2.5 稳定性试验
取S2号样品溶液,在室温下保存,分别于0、2、4、8、24 h测定,结果见表8。由表8可知,各共有峰峰面积的相对标准偏差均小于3%,说明北沙参样品溶液在24 h内是稳定的。
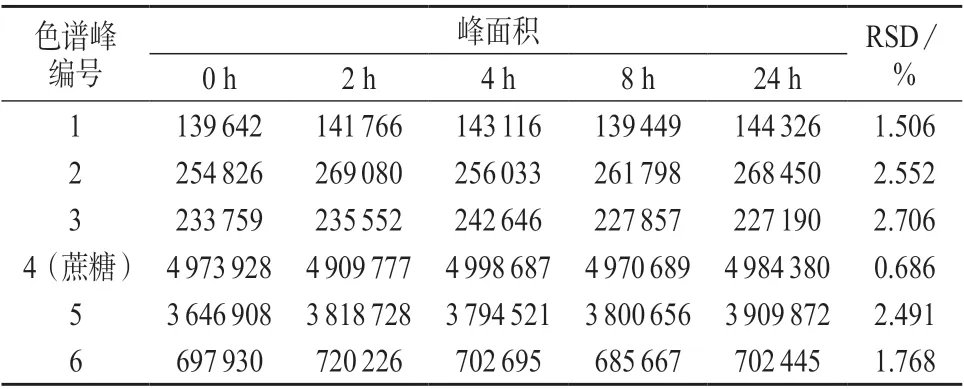
表8 稳定性试验结果
2.6 重复性试验
取S2号样品,按照样品溶液的制备方法,平行制备6份供试液,分别进样,记录HPLC色谱峰面积,计算北沙参样品中的蔗糖含量,结果见表9。由表9可知,测定结果的相对标准偏差均小于3%,说明该方法具有良好的重复性。

表9 ELSD检测器测定的重复性实验结果
2.7 回收试验
取已知蔗糖含量的北沙参粉末约2 g,精密称定,平行6份,分别加入蔗糖对照品约60 mg,按照样品溶液的制备方法制备供试液,进行HPLC测定,结果见表10。由表10可知,平均回收率为98.85%,表明该方法准确度较高。

表10 回收试验结果
2.8 指纹图谱的建立
将对照品溶液和各批北沙参样品溶液分别进样,记录HPLC色谱数据。选择了6个色谱峰为共有峰,以4号峰为参照峰,计算各共有峰的相对保留时间和相对峰面积值,结果见表11、表12。

表11 指纹图谱的共有峰相对保留时间结果
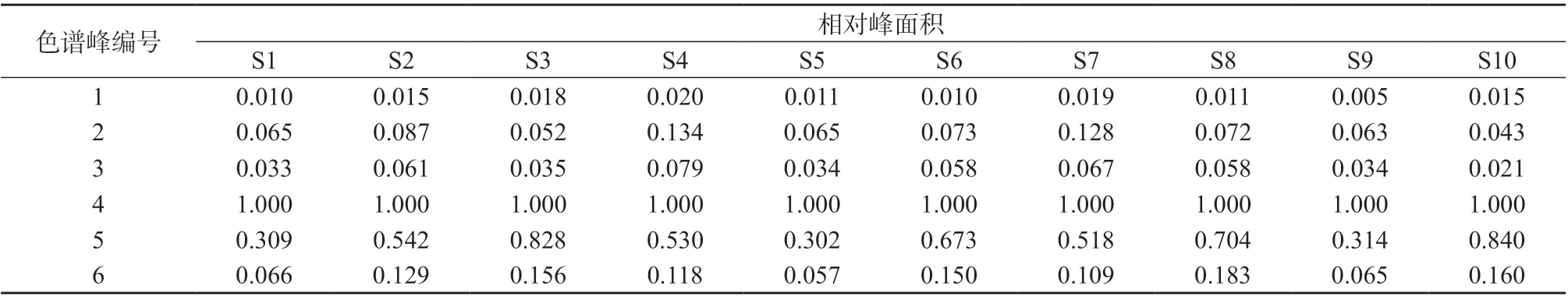
表12 指纹图谱的共有峰相对峰面积结果
2.9 指纹图谱相似度分析
采用国家药典委员会开发的中药色谱指纹图谱相似度评价系统研究版(2004A),对10批北沙参药材的指纹图谱进行相似度分析。该软件具有生成校对照图谱的功能,相似度计算方法为夹角余弦法,可支持多点校正。首先将实验数据导入中药指纹图谱相似度计算软件,选定4号峰为参比峰,设定中位数的匹配模式,将色谱峰进行多点校正后自动匹配,即自动生成对照谱峰,相似度计算结果见表13。由表13可知,与对照指纹图谱相比,10批北沙参药材平均相似度为0.985,相似性结果良好;北沙参药材之间的相似度值为0.917~1.000,说明各批药材间存在差异。因此本实验所建立的HPLC指纹图谱可作为北沙参药材质量控制的有效手段之一。
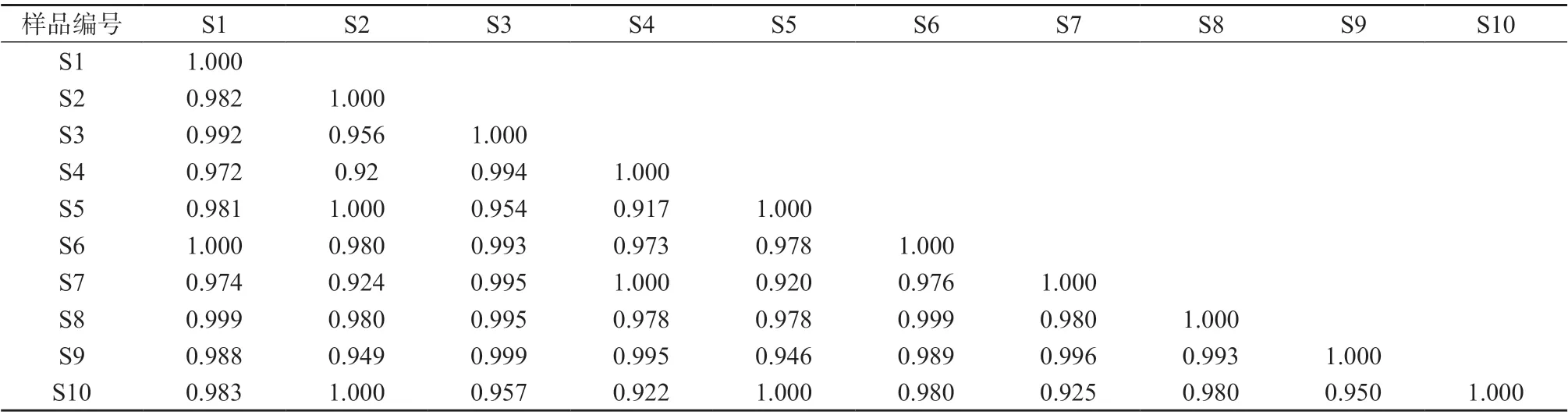
表13 批北沙参药材HPLC指纹图谱相似度计算结果
3 结语
建立了HPLC-ELSD法测定北沙参蔗糖含量的方法,该法测定蔗糖保留时间稳定,测定结果准确、精密度高、回收率符合检测要求。与紫外检测器比较,具有干扰少、基线无漂移、专属性强等特点。建立了北沙参药材的指纹图谱,不同来源北沙参药材之间虽存在一定差异,但整体相似度良好,可作为北沙参药材的质量控制的有效手段之一。2020版药典中并没有对北沙参的含量进行规定,仅通过性状鉴别无法全面的评价药材质量,本研究可以为更好地评价北沙参的质量提供参考。
猜你喜欢
广西糖业(2022年4期)2023-01-03
长春中医药大学学报(2022年11期)2022-12-23
科学大众(2021年18期)2021-10-06
食品与药品(2020年1期)2020-03-10
种子(2019年5期)2019-07-02
中国果业信息(2019年10期)2019-01-05
火力与指挥控制(2018年10期)2018-11-13
浙江工业大学学报(2017年5期)2018-01-22
电子制作(2017年10期)2017-04-18
雷达学报(2015年2期)2015-03-07
