
4-氨基-3,7-二硝基-1,2,4-三唑并[5,1-c]1,2,4-三嗪(TTX)合成机理与性能
2021-06-16 02:57闫峥峰汪营磊陆婷婷赵宝东葛忠学
含能材料 2021年6期
闫峥峰,汪营磊,2,陆婷婷,赵宝东,陈 斌,葛忠学,2
(1. 西安近代化学研究所,陕西 西安 710065;2. 氟氮化工资源高效开发与利用国家重点实验室,陕西 西安 710065)
1 引言
芳香氮杂稠环含能化合物具有较好的爆轰性能、较低的机械感度和较高的热分解温度,近年来受到国内外含能材料学者的关注和研究[1-5],有望开发出新型高能不敏感含能材料。由于芳香氮杂稠环含能化合物分子结构更加紧密,且具有利于分子堆积的近平面结构,密度较高,有效提高了含能化合物的能量水平;同时,离域化的π 电子显著提高了稠环骨架的稳定性,使之热分解温度更高,机械感度降低。
1,2,4-三唑并[5,1-c]1,2,4-三嗪是典型的芳香氮杂稠环骨架,Davin G P 等[6]和Dheeraj K 等[7]分别于2016 年和2017 年先后报道了一种基于该类芳香氮杂稠环的含能化合物4-氨基-3,7-二硝基-1,2,4-三唑并[5,1-c]1,2,4-三嗪(TTX)。TTX 密度为1.82 g·cm-3,略高于RDX,其爆速为8580 m·s-1,爆压为31.2 GPa,生成焓高达403.5 kJ·mol-1,热分解温度为271.8 ℃。文献[6-7]报道TTX 的撞击感度大于60 J,摩擦感度大于360 N,其机械感度远优于RDX,是一种性能接近RDX 的高能不敏感含能化合物。
TTX 是经由5-氨基-3-硝基-1,2,4-三唑(ANTA)重氮化后,与硝基乙腈钠盐偶合环化合成。其中,偶合中间体的环化过程是合成芳香氮杂稠环的关键步骤,但其环化机理尚不明确。此外,作为一种新型含能化合物,TTX 与传统含能组分的相容性尚不明确,需进一步研究。因此,本研究采用密度泛函理论方法(Density Functional Theory,DFT)研究了偶合中间体的环化芳构化过程,并通过实验对偶合中间体的环化过程进行验证,旨在探究TTX 的环化芳构化机理,为此类唑环并三嗪环的构筑提供理论依据。以DFT 计算结果为依据,通过实验研究了pH 对环化过程的影响;采用DSC 法研究了TTX 与推进剂主要组分的相容性,为高能钝感推进剂研制提供物质基础和理论支撑。
2 实验部分
2.1 试剂与仪器
亚硝酸钠,浓硫酸,氯化铵,氢氧化钠,水合肼,浓盐酸(36.5%),高锰酸钾,氢氧化钾,醋酸钠,分析纯,成都市科龙化工试剂厂。5-氨基-3-硝基-1,2,4-三唑(ANTA)参照文献[8-10]合成;硝基乙腈钠盐参照文献[11-13]合成。
NEXUS 870 型FT-IR,美国Nicolet 公司;Vario EL-Ⅲ型元素分析仪,德国Elementar 公司;AV 500 型超导核磁共振仪,瑞士Bruker 公司;Netzsch DSC204HP 差示扫描量热仪,德国Netzsch 公司。
2.2 计算方法
本研究采用了密度泛函理论(DFT)的B3LYP[14-15]的方法在6-31G*[16]基组下,从TTX 的晶体结构出发,对从偶合中间体到TTX 的环化过程的过渡态的几何构型进行全优化,并通过频率分析加以验证。所有计算都采用Gaussian 09 程序包[17]进行。
2.3 实验过程
TTX 的合成路线如Scheme 1[7]所示。

Scheme 1 Synthetic route of TTX[7]
将ANTA 1.29 g(0.01 mol)和2.5 mL 浓HCl 加入10 mL 水中,加热溶解后冰盐浴降温至-5 ℃,将0.76 g NaNO2(0.011 mol)溶解于10 mL 的冰水中,滴加入ANTA 的盐酸溶液中,滴加完毕后-5~0 ℃反应0.5 h,将硝基乙腈钠溶液滴加入该反应液,5 ℃以下反应2 h,室温反应4 h。旋蒸浓缩后过滤,冷水洗两次,将固体溶解于20 mL 甲醇与水的混合溶剂(1∶1)中,调pH 至3~4,70 ℃反应4 h,冷却过滤,冷水洗两次,晾干得淡黄色固体1.52 g,总产率67.3%。1H NMR(DMSO-d6,500 MHz)δ:10.90(s,1H),10.19(s,1H);13C NMR(DMSO-d6,125 MHz)δ:163.9,155.9,140.5,139.5;FT-IR(KBr,ν/cm-1):3440,3414,3312,3292,3226,1656,1566,1495,1476,1456,1439,1408,1333,1290,1252,1210,1178,1091,1006,842,778,770,757,704,677,633,585,532;Anal.Calcd for C4H2N8O4(%):found(calculate value),C 21.31(21.25),H 0.90(0.89),N 49.71(49.56)。
3 结果与讨论
3.1 TTX 环化机理DFT 分析
将ANTA 的重氮盐与硝基乙腈偶合后,得到中间产物R,R 通过亲核加成经过渡态TS 得到中间产物P1,再经历芳构化重排得到目标产物P2(TTX),反应历程及原子标示如Scheme 2 所示。

Scheme 2 Possible mechanism of cyclization
由中间产物R 环合成目标产物TTX 的可能机理如下:类吡咯氮原子N(2)作为亲核中心对氰基的碳原子C(3)进行亲核加成,H(1)与N(4)成键得到过渡态TS,进而N(2)与C(3)成键得到中间体P1,P1 经过芳构化重排得到共轭范围更大的芳香稠环结构目标产物P2(TTX)。本研究根据Scheme 2 所示可能的反应途径,采用Gaussian 09 程序包在B3LYP/6-31G*水平下计算了环化过程的过渡态能垒,结果如图1 所示,其中过渡态TS 只有唯一的虚频468.3i cm-1,中间产物R、中间体P1 和产物P2 均无虚频存在。从图1 中可以看出,从中间产物R 需要较高的活化能(155.8 kJ·mol-1)才能得到过渡态TS,在这一过程中,N(2)—H(1)的距离从0.1017 nm 增加到0.1875 nm,而H(1)—N(4)的距离从0.2087 nm 减小到0.1059 nm,表明三唑环上类吡咯氮原子上的H(1)转移到了氰基的氮原子N(4)上。在从过渡态TS 到中间体P1 的过程中,N(2)—C(3)的距离从0.2826 nm 缩短到0.1424 nm,证实在这一过程中形成了新的N—C 键,且这一过程的自由能变化为-208.0 kJ·mol-1,是一个较快的自发过程。在从中间体P1 转化为产物P2 的过程中,C(3)—N(4)的距离从0.1184 nm 增加到0.1264 nm,这是芳构化过程中,C(3)与N(4)间从双键转化为单键,这一过程的自由能变化为-41.7 kJ·mol-1,为自发过程。
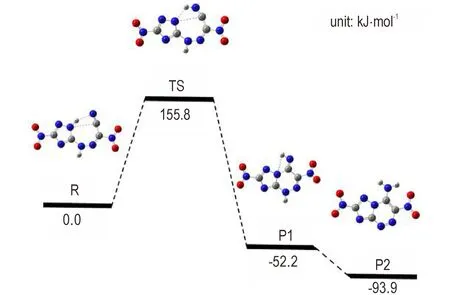
图1 偶合中间体到TTX 环化过程的能量曲线Fig.1 Energy profile of cyclization process of coupling intermediate to TTX
从计算环化过程的过渡态、中间体的构型和能垒变化可以看出:(1)从偶合中间产物R 到过渡态TS 过程的能垒较高,达到155.8 kJ·mol-1,说明反应需要较长的反应时间或较高的反应条件;(2)在这一过程从H(1)—N(2)键的断裂,形成H(1)—N(4)键开始,得到过渡态TS,需要氢原子的参与;(3)过渡态TS 到中间体P2 的过程中,自由能变化为-208.0 kJ·mol-1,是一个较快的自发过程;(4)从中间体P1 到产物P2 的芳构化过程的自由能变化为-41.7 kJ·mol-1,是一自发过程。
3.2 TTX 环化过程
研究了反应温度和体系pH 对偶合中间体R 环化过程的影响,发现以下实验现象佐证了理论计算分析推论:(1)偶合中间体R 环化过程需要在室温条件下反应3 d,或者70 ℃下反应3~4 h,较慢的环化反应速率说明引发反应所需的活化能较高,这与计算结果一致;(2)通过研究不同pH 条件下环化反应发现,在中性或弱碱性(pH=7~8)的条件下,偶合产物室温下反应3 天或70 ℃下反应3~4 h 均不能得到产物。在酸性(pH=3~4)条件下,相应的环化条件下可得到产物。研究发现,碱性条件下中间体R 的H(1)被夺走,生成化合物RS(化合物RS 与化合物R 的13C NMR 对比如图2 所示,从图2 中可以看出,相比于化合物R,化合物RS 的碳谱高场区硝基乙腈模块的碳原子的化学位移基本保持一致,1,2,4-三唑环上的碳原子明显向低场区移动,这是由于成盐后芳香环的电子云密度增大,去屏蔽效应增强,导致相关碳原子的化学位移向低场移动)。由于缺少氢原子H(1),不能对氰基氮原子N(4)进行加成,故反应无法进行。这一结果与上述的化学键的变化相符。

图2 化合物R 和RS 的13C NMR 化学位移,ppmFig.213C chemical shifts of compounds R and RS,ppm
3.3 TTX 热性能
3.3.1 TTX 热分解
在氮气气氛下,在50~375 ℃温度区间以10 ℃·min-1为升温速率测试了TTX 的DSC 曲线,以研究TTX 热分解过程,结果见图3。从图3 中可以看出在TTX 只有一个放热峰,表明TTX 的热分解只有一个过程,热分解峰温为281.8 ℃。在同样的测试条件下,RDX 的热分解峰温为227.5 ℃[17],说明TTX 比RDX 具有更好的热稳定性。TTX 的热分解起始温度为279.3 ℃,终止温度为286.1 ℃,热分解温度范围较窄,说明TTX 具有较快的热分解速率,有利于提高推进剂的燃速。
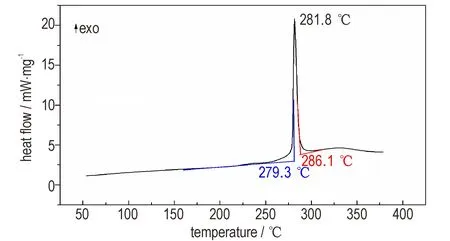
图3 TTX 的DSC 曲线Fig.3 DSC curves of TTX
3.3.2 TTX 热分解过程的活化能
为进一步分析TTX 的热分解过程,采用非等温DSC 法计算了TTX 的热分解表观活化能,结果见图4。由图4 可知,TTX 在相应的升温速率下的热分解峰值温度分别为272.1,277.3,281.8 ℃和286.4 ℃。
随后,采用Kissinger 方程(式1)和Ozawa 方程(式2)获得TTX 的热分解动力学参数。通过Kissinger方程和Ozawa 方程获得的TTX 的热分解表观活化能分别为361.2 kJ·mol-1和352.2 kJ·mol-1,二者比较接近。且两者方法的线性相关系数r 都超过0.99(Kissinger 方程r=0.9983,Ozawa 方程r=0.9984)。

图4 不同升温速率下TTX 的DSC 曲线Fig.4 DSC curves of TTX at different heating rates
Kissinger 方程[18]:

Ozawa 方程[19]:

式中,β 为升温速率,K·min-1;Tp为热分解峰温,K;R 为理想气体常数,8.314 J·mol-1·K-1;Ek和Eo分别为Kissinger 方程和Ozawa 方程计算得出的表观活化能,J·mol-1;A 是指前因子,C 为常数。
由TTX 非等温热分解动力学研究可以看出,TTX的热分解表观活化能高于TATB(211.4 kJ·mol-1)[20],说明TTX 对热刺激敏感性较低,这可能得益于TTX 较大的共轭结构。
3.3.3 TTX 与常用含能材料的相容性
通过差示扫描量热法(DSC),参考国军标GJB772A-1977 502.1 方法评价TTX 与常用含能材料HMX、RDX、Al 和硝化棉(NC)的相容性,评价的标准或判据为混合体系与原组分两者的DSC 热分解峰温Tp之差ΔTp。ΔTp=Tp2-Tp1,式中,Tp1为原组分的热分解峰温,为混合体系的分解峰温。当ΔTp为:0~2℃时,混合体系相容;3~5 ℃时,混合体系轻微敏感,可短期使用;6~15 ℃时,混合体系敏感,避免混合使用;大于15 ℃时,混合体系危险,禁止混用[21-22]。实验测试条件为:氮气气氛,气体流速50 mL·min-1,升温速率10 ℃·min-1,样品量1 mg 左右。
将TTX 与等质量的HMX、RDX、Al 粉、NC 混合后,其DSC 曲线如图5 所示。从图5a TTX 与HMX 和RDX 的DSC 曲线中可以看出,TTX 与HMX 的热分解峰温很接近,二者混合后热分解放热峰只有一个,为277.2 ℃,分别比TTX 和HMX 的热分解峰温提前了4.6 ℃和5.3 ℃,表明TTX 与HMX 之间存在一定的相互作用,轻微敏感。TTX 与RDX 的混合物只有一个放热峰,峰温239.3 ℃,并在接近RDX 分解峰的温度附近有一个肩峰,说明RDX 分解后促进TTX 的分解,使TTX 提前分解完全。从图5b 中可以看出,TTX 与Al 混合后,热分解峰温提前了1.3 ℃,表明二者无明显相互作用,具有较好的相容性。TTX 与NC 混合后,NC 热分解过程基本没有变化,TTX 的分解峰温提前了5.3 ℃,说明NC 对TTX 的热分解历程有一定的促进作用。相容性研究表明:TTX 与Al 粉相容性良好;与HMX 有一定相互作用,轻微敏感;RDX、NC 会明显促进TTX 热分解,混合体系较为敏感,应避免混合使用。

图5 TTX 与HMX,RDX,NC,Al 及其混合体系的DSC 曲线Fig.5 DSC curves of TTX,HMX,RDX,NC,Al and their mixed systems
3.4 TTX 安全性能
采用排气法测试了TTX 的密度,通过BAM 撞击感度测试仪测试了TTX 样品的撞击感度,将TTX 与RDX的热性能和机械感度等对比于表1。
从表2 中可以看出,TTX 密度高于RDX,生成焓高达403.5 kJ·mol-1,明显高于RDX。TTX 实测热分解峰温为281.8 ℃,远高于RDX。TTX 的撞击感度为60 J,摩擦感度大于360 N,其机械感度远优于RDX。总体来说,TTX 是一种爆轰性能接近RDX,生成焓较高,热性能和机械感度优于RDX 的含能材料。

表1 TTX 与RDX 性能对比Table 1 Properties of TTX and RDX
TTX 的性能优势可能得益于其特殊的结构因素:首先,氮杂稠环结构紧凑,使之密度较高,进而具有较优的爆轰性能;其次,三唑并三嗪的稠环芳香性一方面通过提高稠环骨架的稳定性而提高了化合物的热分解温度,另一方面,芳香性使得稠环内的原子尽可能处于同一平面,且通过共轭效应减小环外硝基、氨基与母环的扭转角,进而使得TTX 具有类平面结构,即有利于分子紧密堆积提高密度,又利于降低感度;最后,硝基乙腈在三嗪环外引入了相邻的氨基和硝基,核磁氢谱中氨基峰分裂成双峰和晶体学结构数据(氨基氢与相邻的硝基上的氧原子距离为2.13 Å[6])都证实形成了分子内氢键,有利于感度降低。可以看出,氮杂芳香稠环是平衡含能材料能量与稳定性的重要结构因素,硝基乙腈是合成邻硝基氨基三嗪类稠环的重要试剂,应进一步探索氮杂芳香稠环含能材料的合成技术,实现含能材料能量性能与安全性能的同步提升。
4 结论
(1)采用密度泛函理论研究了TTX 合成过程中偶合中间体的环化芳构化过程,通过能垒分析发现环合过程首先是类吡咯氮原子作为亲核中心对氰基碳进行亲核加成反应,氢原子由类吡咯氮原子上转移到氰基氮上,得到过渡态TS,进而得到中间体P1,P1 经过芳构化重排得到共轭范围更大的芳香稠环结构目标产物P2。该环化过程的能垒较高,环化需要较长的实验时间或较高的反应温度;环化需要在酸性条件下进行,以满足环化过程氢原子转移的需要。
(2)TTX 的热分解温度为281.8 ℃,热分解表观活化能为356.7 kJ·mol-1,是一种热稳定性较高的含能材料;相容性研究表明TTX 与Al 相容,与HMX 有一定相互作用,轻微敏感,而RDX、NC 会明显促进TTX 热分解,混合体系较为敏感,应进一步研究其相互作用。
(3)TTX 的爆速为8580 m·s-1,爆压为31.2 GPa为,与RDX 基本相当;生成焓为403.5 kJ·mol-1,撞击感度为60 J,摩擦感度大于360 N,均优于RDX,是一种高能不敏感化合物。
猜你喜欢
佳木斯大学学报(自然科学版)(2022年1期)2022-11-25
北京航空航天大学学报(2022年5期)2022-06-06
中国药学药品知识仓库(2022年10期)2022-05-29
汕头大学学报(自然科学版)(2020年4期)2020-12-14
火炸药学报(2020年5期)2020-10-27
电脑知识与技术(2018年3期)2018-03-21
兵器装备工程学报(2017年10期)2017-11-14
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
股市动态分析(2015年12期)2015-09-10
