
基于转录组数据识别结直肠癌的特异性功能模块*
2021-06-07 05:41陈晓琳许德华廖苑君李让孙胜南蓝树金饶绍奇
肿瘤预防与治疗 2021年4期
陈晓琳,许德华,廖苑君,李让,孙胜南,蓝树金,饶绍奇
523808 广东 东莞,广东医科大学 公共卫生学院(陈晓琳、许德华、廖苑君、李让、孙胜南、蓝树金),医学系统生物学研究所(陈晓琳、许德华、廖苑君、李让、孙胜南、蓝树金、饶绍奇)
结直肠癌(colorectal cancer,CRC)是常见的消化道恶性肿瘤,为全球第4大致命的癌症(仅次于肺癌、肝癌和胃癌),每年约有90万人死于该疾病[1]。近年来,CRC的发病率和死亡率呈上升趋势,主要原因是现代人饮食习惯和生活方式的改变。据预测,到2030年,全球CRC负担将增加60%,新增病例将超过220万,癌症死亡将超过110万[2]。目前,改善CRC的治疗效果逐渐成为人们关注的重点。尽管CRC的新疗法取得了一定成效,包括原发性疾病的腹腔镜手术,转移性疾病(如肝和肺转移)的切除术,放射治疗与新辅助治疗、姑息化疗[3]。然而,这些新疗法对提高CRC的治愈率和长期生存率还存在一定的局限性,CRC的5年生存率仍很低。因此,揭示CRC的病因与发病机制对其预防和治疗具有重要意义。
近年来,大量研究利用生物信息学方法在分子水平上进行数据挖掘,为研究各种疾病的致病机制提供新思路。迄今为止,分析基因表达谱数据已被证明有助于更好地探究疾病的发病机制、辅助临床诊断和判断药物疗效,尤其在癌症中已经确定了许多生物学过程和疾病的分子基础[4]。为此,本研究通过收集GEO数据库中CRC芯片数据集,利用大规模的转录组数据分析与CRC致病相关的特异性功能模块和核心基因,并对各功能模块进行通路富集分析,为进一步阐明CRC的病因、发病机制及预后提供重要依据。
1 材料与方法
1.1 数据来源
从基因表达综合GEO数据库(https://www.ncbi.nlm.nih.gov/gds/)中按照以下标准获取CRC的基因芯片数据集:1)以“colorectal”为关键词检索与CRC相关的转录组基因芯片;2)选择物种为“Homo sapiens”、研究类型为“expression profiling by array”两个过滤条件进一步筛选数据;3)最后选定实验设计符合病例对照研究类型并且病例组织样本数均大于30的基因芯片。最终获取CRC的4个基因芯片数据集:GSE44076(98病例+50对照)、GSE21815(132病例+9对照)、GSE9348(70病例+12对照)、GSE8671(32病例+32对照)。
1.2 筛选差异性表达基因
本研究利用在线分析工具GEO2R(http://www.ncbi.nlm.nih.gov/geo/geo2r/)分别筛选4个芯片数据集的差异表达基因(differentially expressed genes,DEGs),条件设定为校正P值小于0.05且logFC的绝对值大于1。然后运用在线工具jvenn(http://jvenn.toulouse.inra.fr/app/example.html)对4个芯片数据集的上下调的DEGs进行取交集,得到4个芯片数据集的上下调DEGs的交集。
1.3 构建特异性基因网络
HPRD数据库[5](http://www.hprd.org/)是人类蛋白质参考数据库,其中包含人类蛋白质互作(protein-protein interaction,PPI)注释信息。当前,该数据库共收集了41 327条PPI对子数据,是目前关于人类蛋白互作数据可信度最高的数据库之一。
本研究假设初始种子基因为筛选得到的CRC共同差异基因。而CRC致病基因网络的节点集合主要包含两类基因,初始种子基因及其在PPI的一级邻居基因。对于集合中的任意2个不同基因,只要该基因产物在PPI网络中存在互作关系,则视它们在网络中存在连接。基于以上假设,我们构建了一个无向的CRC特异性基因网络,其中所有节点表示基因,节点之间的边表示基因间存在互作关系。最终,通过Cytoscape软件(版本3.6.1)实现PPI网络的可视化。
1.4 挖掘网络的特异性功能模块
蛋白质相互作用网络中存在具有统计学意义的模块,对这些模块进行分析不仅能够在一定程度上降低网络的复杂性,同时能够对具有生物学意义的有效信息进行提取。功能模块在通常情况下能够被视为功能相似且较为独立的子结构或子网络,同一个模块中的蛋白质互相之间有较显著的功能性。因此,同随机网络比较而言,模块化网络的连接将表现出更优异的紧密性。通过功能模块的角度对网络进行分析具有较高的可靠性,同时功能模块的识别实际上是网络结构分解的优化问题。本研究应用Newman算法[6-7]对CRC基因网络展开一分为二的循环式网络分解,识别网络中高度模块化的功能模块。该算法的基本思想在于将网络分解问题转变成二次型优化问题,即求使目标函数Q最大的列向量s所对应的数值:
在方程式中s包括两类基因,分别是初始种子基因及其在PPI中的一级邻居基因,A为集合s的邻接矩阵(adjacent matrix),通常视为网络中基因间的相互连接关系,其中Aij=1表明基因i和基因j存在互作关系,相反记为Aij=0;m为网络中边的总数,ki为节点i的连通度,kj为节点j的连通度。因此s是取值1或-1的指示列向量,si为基因i所属的子网,则:
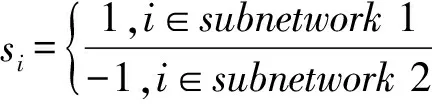
根据柯西(Cauchy)不等式,若s取模块化矩阵B的最大特征值对应的特征向量,那么目标函数Q则最大。由于s只取±1值,故s仅会保留特征向量对应元素的正负号,同时结合符号方向,将网络分为两个部分。对上述网络一分为二的过程重复操作,一直持续到无法分解为止,最后我们将节点数大于60的子网作为模块。
1.5 功能模块拓扑学属性分析
本研究从以下4个指标进行功能模块的拓扑学属性分析:
1)连通度(degree):指网络中直接与某一节点v相连的边的数目。连通度是描述单一节点的最基本的拓扑性质,通常把连通度较大的中心节点(hub)作为研究的重点。因此,该指标的值能够较为准确地反映出网络中节点的重要性,可以看作获取网络中流动性的程度。某个节点的连通度大于或等于t的概率为:
在方程式中λ=NP,λ为节点的期望连通度,N为节点数目,P为任意两个不同节点发生连接的概率。对核心节点进行泊松分布检验,设定显著性水准Bonferroni校正后P<0.01。
2)网络直径:指网络中任意两节点间最短路径的最大值。它是描述网络总体性质的属性,可反映网络的大小。
3)特征路径长度:指网络中所有节点间的最短路径的平均值,可反映模块的紧密程度。
4)无标度网络:指网络中连通度的分布符合幂率分布,即p(k)∝k-α,其中k为接通度,α为指数参数,可采用极大似然法估计[8]。幂率分布表明在无标度网络中少数连通度很大的关键节点控制着整个网络,使网络紧密地连接在一起。本研究通过Kolmogorov-Smirnov(KS)拟合优度检验判断网络的无标度特性。
1.6 KEGG通路富集分析
采用R语言(版本3.5.2)的“cluster profiler”软件包对每个功能模块进行京都基因与基因组大百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)生物学通路富集分析。设定显著性水准FDR(false discovery rate)校正后P<0.001,对各功能模块前5条通路开展后续分析。通过Cytoscape软件实现功能模块与通路分类的关系的可视化。本研究按照上述方法步骤进行,具体技术路线见图1。
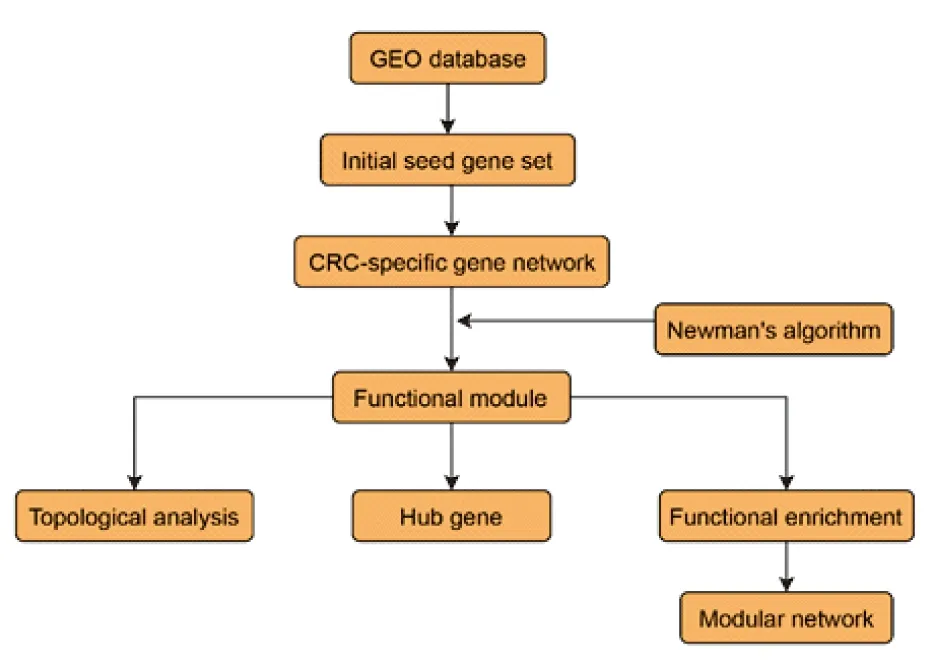
图1 数据分析流程图
2 结 果
2.1 筛选差异性表达基因
通过在线工具GEO2R对4个芯片数据集进行差异表达分析。GSE44076数据集经筛选后得到3 160个DEGs;GSE21815数据集经筛选后得到8 528个DEGs;GSE9348数据集经筛选后得到4 914个DEGs;GSE8671数据集经筛选得到3 411个DEGs。通过在线工具jvenn得到4个数据集的交集基因,其中包括412个上调DEGs以及231个下调DEGs。4个数据集的上下调基因韦恩图见图2。
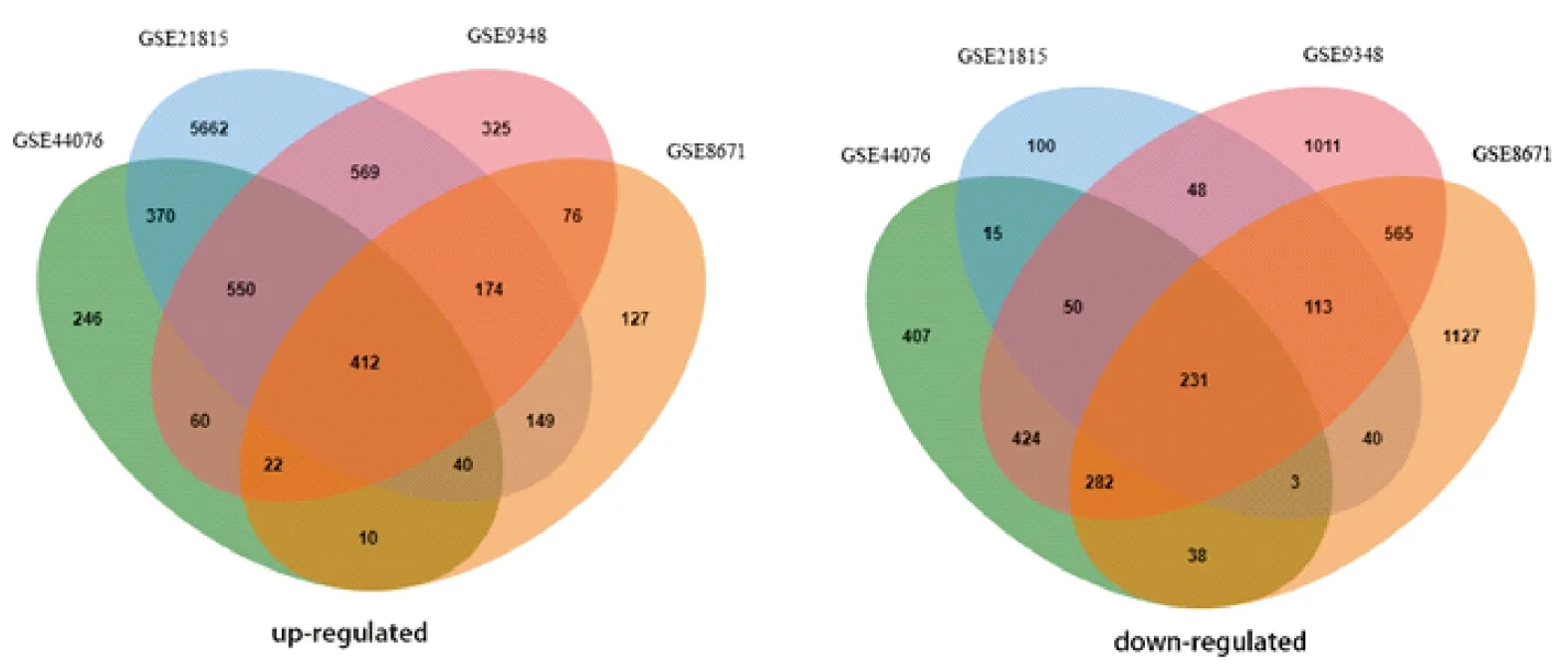
图2 差异表达基因的韦恩图
2.2 构建特异性基因网络及拓扑学属性分析
本研究利用HPRD数据库构建CRC特异性基因网络,网络中包含所有差异基因的互作关系。原始网络含有2 460个节点和3 576个互作对子,节点代表基因,边代表蛋白互作。随后删除孤立作用的节点(SST、GTF3A、1L1R2等),最终生成的最大子网络含有2 261个节点和3 450个互作对子。通过Cytoscape在线分析工具NetworkAnalyzer对最大子网络进行拓扑属性分析,结果显示聚类系数为0.034,网络直径为17,网络平均路径长度为3.048,特征路径长度为5.428。将最大子网络中连通度排名前10的核心节点进行拓扑学属性分析(表1)。其中经Pubmed数据库检索发现,目前尚未有文献明确报道PRKCB、KAT2B、ACTN1等基因在CRC发生发展中的作用。因此,这些基因可能是临床上诊治CRC的潜在靶点。
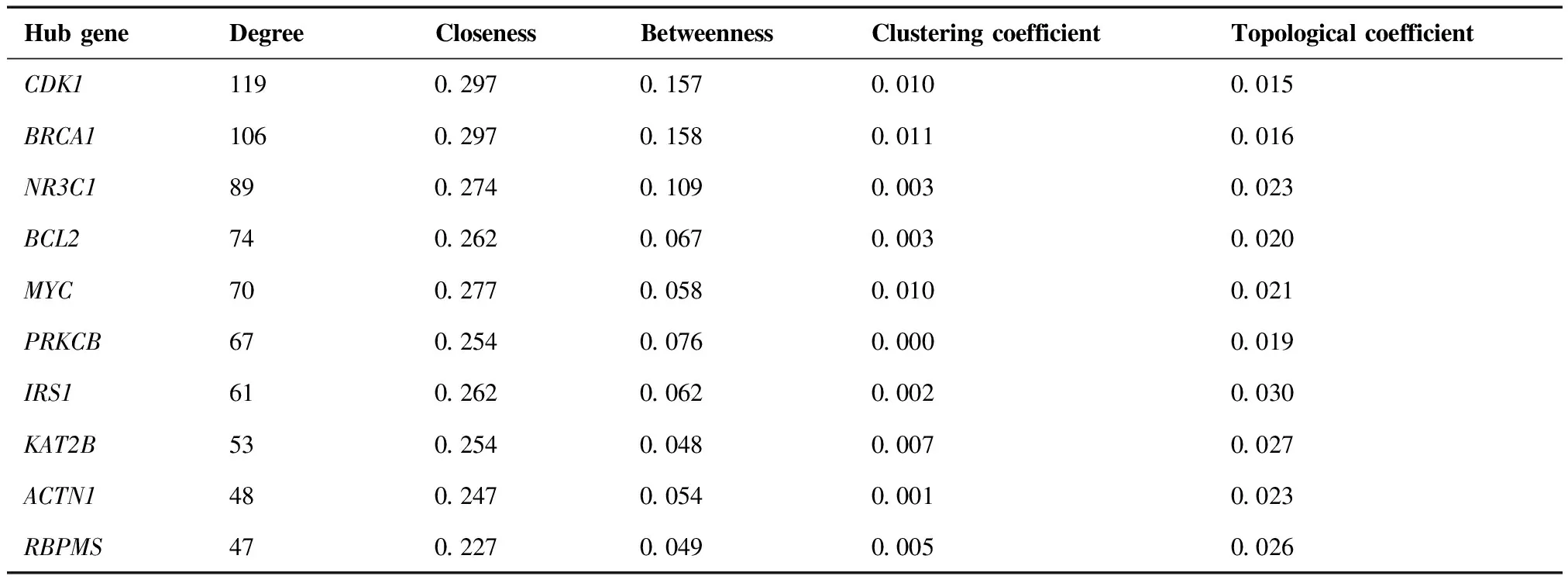
表1 连通度排名前10的核心节点的拓扑学属性
2.3 挖掘网络的特异性功能模块
应用Newman算法挖掘分解CRC的最大子网络,将15个节点数大于60的功能模块纳入后续分析。15个功能模块的基本特征和拓扑属性见表2。从表2可见,不同功能模块的节点数有较大区别,本研究最大的模块(M11)包含352个基因(其中初始种子基因有47个)和557条边,平均每个节点有1.6条边。最小的模块(M13)包含60个基因(其中初始种子基因有9个)和61条边,平均每个节点有1条边。这些功能模块的拓扑学特征相似并符合“六度分离理论”[9],它们的网络直径波动范围在7~14,特征路径长度均小于6。各功能模块的无标度性可从两方面体现。首先结果显示多数功能模块指数参数α在2~3之间,符合生物学无标度网络的特征。同时KS拟合优度检验结果可见各功能模块的连通度均近似服从幂率分布(P>0.05),可判断为无标度网络。
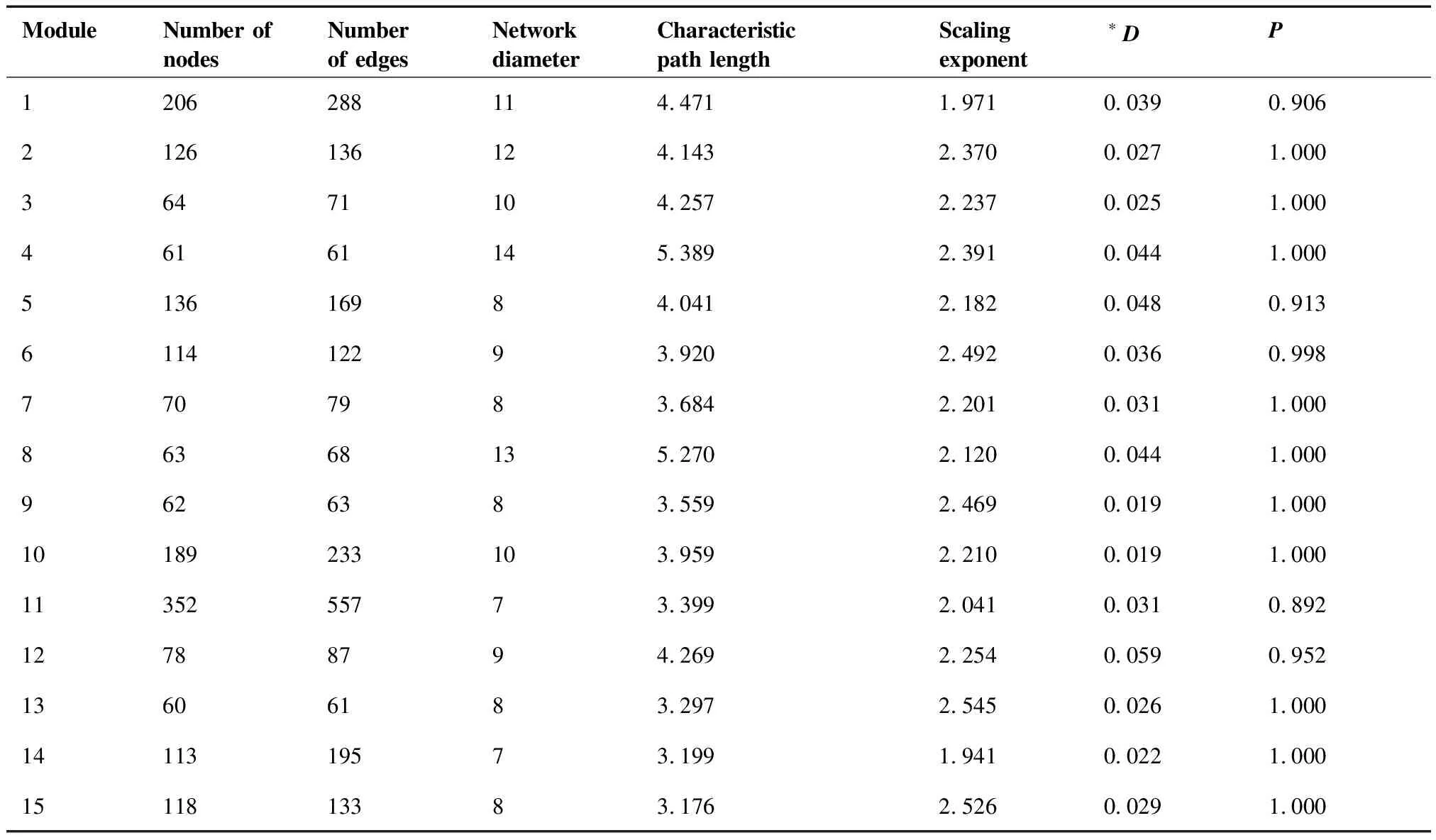
表2 各模块的基本特征和拓扑属性
2.4 识别核心基因
本研究对模块中的基因进行泊松分布检验,设定显著性水准为Bonferroni校正P<0.01。我们共识别了87个核心基因,其中通过PPI发现的核心基因有4个(LPL、NR3C1、ETS2、TP53),列出各个功能模块的核心基因(表3)。15个功能模块中,模块11挖掘的核心基因最多:共22个(包含MSX1、CDC25A、TOP2A等),模块3挖掘的核心基因最少:1个。本研究经过R语言软件的“igraph”包实现功能模块与核心基因分布图的可视化(图3)。
通过Pubmed数据库的检索,发现有26个核心基因暂无文献明确报道与CRC相关,这些基因可能成为CRC潜在的分子标志物,例如核受体亚家族5A组成员2 (nuclear receptor subfamily 5 group A member 2,NR5A2)、端粒酶逆转录酶(ETS proto-oncogene 2,ETS2)、丝氨酸蛋白酶抑制H族成员1(serpin family H member 1,SERPINH1)、磷酸肌醇-5-磷酸酶D(inositol polyphosphate-5-phosphatase D,INPP5D)等基因。另外有部分核心基因虽然被报道与CRC相关,但仍然缺乏大量的实验来探究其影响CRC的发病机理,因此着重关注这些核心基因可能为疾病的临床诊治提供参考。例如层粘连蛋白亚基α1(laminin subunit alpha 1,KAMA1)、丝氨酸蛋白酶抑制剂E族成员1(serpin family E member 1,SERPINE1)等蛋白的编码基因。
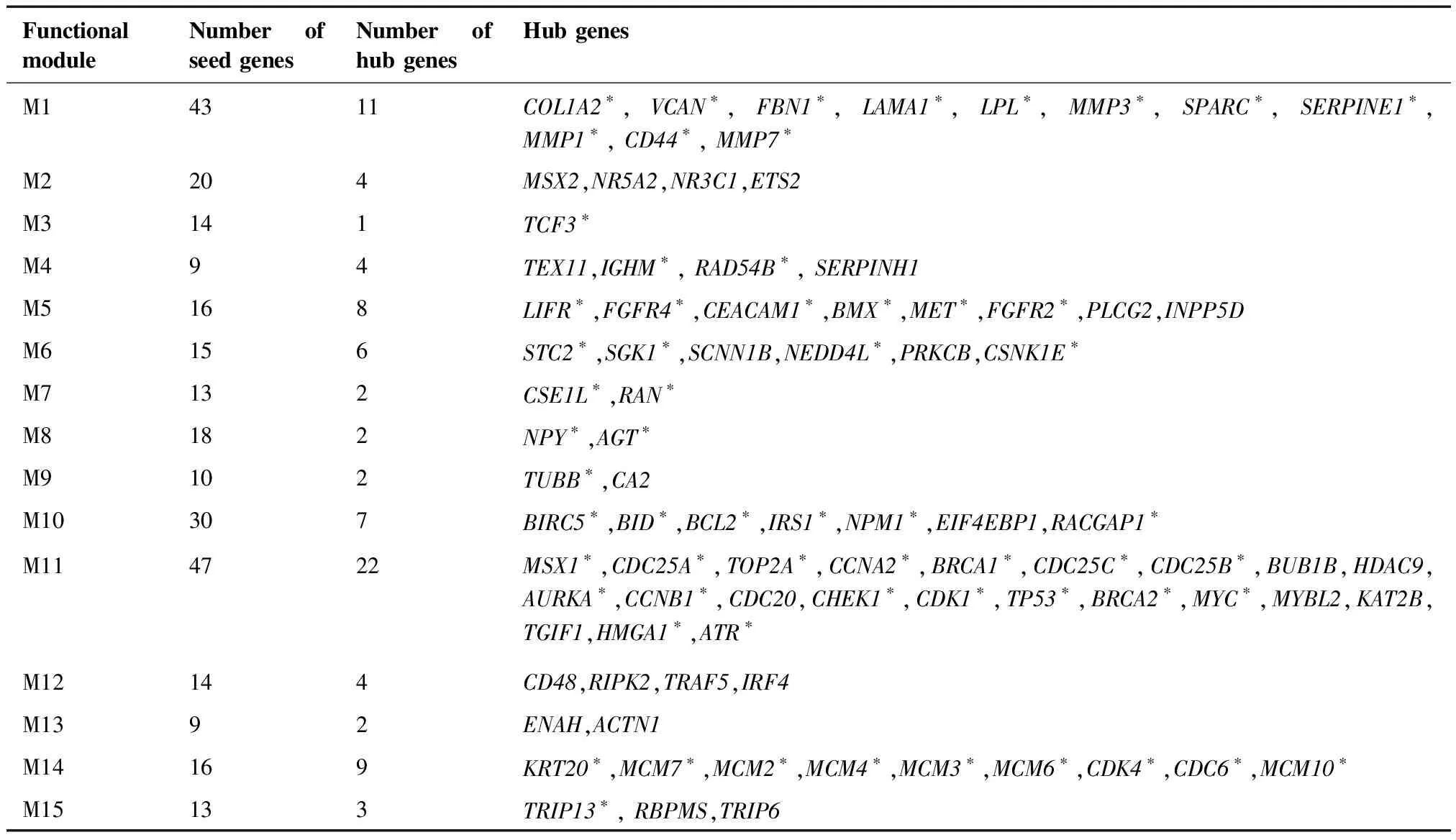
表3 各功能模块的核心基因
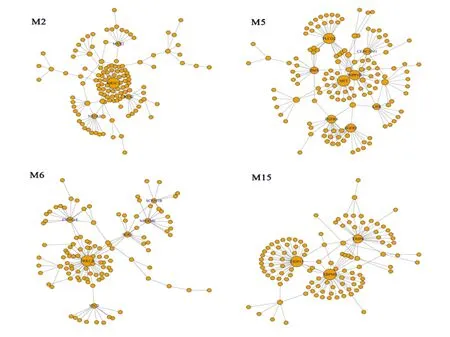
图3 部分功能模块和核心基因
2.5 模块的功能富集分析
15个CRC功能模块经过KEGG通路富集分析后的结果见表4,表4结果显示:15个功能模块有13个模块至少富集到1条生物学通路(除模块M12、M13)。DEGs显著富集于细胞周期(hsa04110)、DNA复制(hsa03030)、Wnt信号通路(hsa04310)、PI3K-AKT信号通路(hsa04151)等有统计学意义的生物学通路(FDR<0.001)。
本研究整合全部富集通路的类别与13个功能模块的相互关系,并对其产生的相互作用进行评估。最终,利用Cytoscape软件实现模块和通路分类关系的可视化(图4)。通路分类结果显示:信号转导(M2、M5、M6、M8)、信号分子与相互作用(M1、M3、M8)以及内分泌系统(M8、M10、M11)、细胞生长与死亡(M10、M11、M14)这4种功能类别的连通度较高,提示CRC的发生与这些功能密切相关,各模块之间联系紧密共同影响疾病的发生发展。
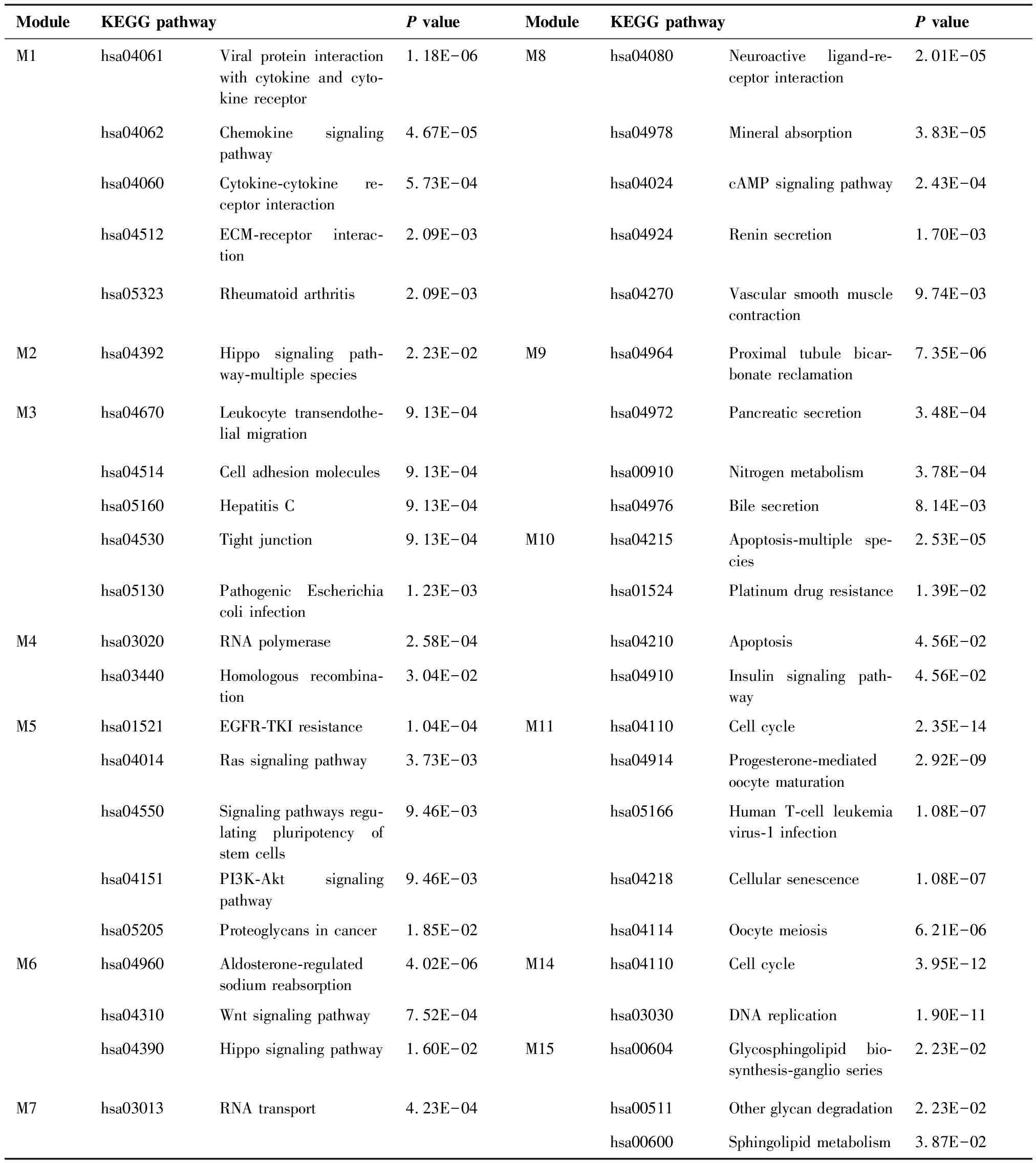
表4 各功能模块的KEGG通路富集结果
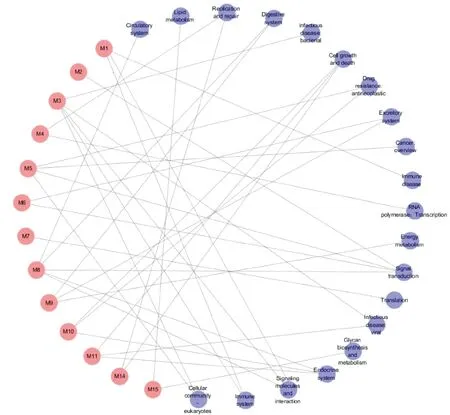
图4 CRC特异性模块与通路功能分类的相互联系
3 讨 论
CRC的复杂进展是一个多因素影响、多阶段作用的过程,是多基因及多条信号通路共同参与的结果。从多基因水平研究肿瘤基因表达谱可能为更好地探索肿瘤发病机制提供依据。虽然近年CRC的研究取得了进展,但它在全球的发病率和死亡率仍居高不下,早期诊断困难、肿瘤易转移复发、5年生存率低等问题仍存在。近年来,基于基因表达数据的分析研究已经确定了许多人类癌症不同类型的基因生物标记物。例如,研究发现BRCA1和BRCA2基因的突变导致了60%的遗传性乳腺癌和卵巢癌病例[10-11]。因此,挖掘与CRC致病相关的核心基因和功能模块,寻找新的潜在基因生物标记物将有助于为CRC开发更有效的诊断和治疗方案。
本研究应用生物信息学方法筛选与CRC相关的412个上调DEGs以及231个下调DEGs,通过PPI知识扩充构建致病基因网络,应用Newman算法提取了15个与CRC相关的功能模块。然后根据连通度分布情况和泊松分布来确定核心基因,共识别出87个核心基因。通常,探讨模块中核心基因的功能有助于揭示疾病发生的病因和发病机制。通过检索Pubmed数据库,发现有一部分基因已被文献报道直接或间接与CRC相关,例如CRC差异表达下调的基因COL1A2以及差异表达上调的基因CDK1、MYC等。许多研究表明COL1A2参与恶性肿瘤的发生和发展。COL1A2的差异表达可能在人类恶性肿瘤中引起明显的胶原介导效应。Kalmar 等[12]发现COL1A2基因高度甲基化,其mRNA表达在CRC样本中显著降低。Yu等[13]报道证实,原发性CRC组织中COL1A2表达明显下调,其mRNA在CRC组织中的表达与肿瘤分化程度、浸润深度、淋巴结转移呈正相关,这些结果提示COL1A2可能是鉴别CRC组织和非恶性组织的潜在生物标志物。另外,有相关研究发现COL1A2在某些恶性肿瘤中具有抑制作用,结合COL1A2过表达会抑制CRC细胞增殖、迁移和侵袭的现象,提示COL1A2在CRC中具有抗癌作用[13-16]。网络中连通度最高的基因是CDK1,它维系着网络中15.70%的基因间最短路径的连通路径。CDK1是G2-M检查点的重要调控因子。有研究表明,CDK1和iASPP在CRC组织和细胞系中表达上调,CDK1蛋白与iASPP蛋白存在相互作用,它们通过p53凋亡途径影响CRC细胞的增殖和凋亡[17]。基于CDK1在癌细胞中的高表达,其不仅可以作为CRC治疗的靶点,而且可以作为一个有效预后指标。MYC是Wnt信号通路的一个靶基因,Wnt/β-catenin信号通路的激活通过解除对包括c-Myc原癌基因在内的下游靶基因的表达调控来驱动CRC的生长。有研究证明敲除MYC或MYC靶向嘧啶合成基因(CAD、UMPS和CTPS)可以抑制CRC细胞增殖[18]。因此,MYC靶向嘧啶合成途径可能是CRC治疗的潜在靶点。
本研究发现一些在网络中连通度较高的基因,尚无文献报道证实其与CRC相关,有进一步探索的价值。例如CRC下调的差异表达基因PRKCB、KAT2B以及上调的差异表达基因ACTN1。通过查阅相关文献发现这些核心基因曾被报道与其他肿瘤的发生发展有关。例如,PRKCB是蛋白激酶C基因家族成员的一个基因[19]。PKC家族成员具有独特的表达趋势,被认为在肿瘤相关过程中起着独特的作用[20-21]。PKCβ1是由PRKCB编码的蛋白,相关研究发现PKCβ1在胰腺PANC1细胞中的重要作用,发现它可抑制胰腺癌的发生[20]。Liu等[22]发现PRKCB在过氧化物酶体增殖物激活受体途径发生遗传变异,可能会导致胰腺癌的易感性。近年来研究发现新型环状RNA hsa_circ_0092306通过调节MKN-45细胞中miR-197-3p/PRKCB的途径促进了胃癌的发展[23]。KAT2B是一种蛋白质编码基因,在肿瘤组织中它的mRNA和蛋白质表达异常。有相关文献报道,与癌旁组织相比,KAT2B在食管鳞状细胞癌[24]、肠型胃癌[25]和肝细胞癌[26]癌组织中的表达下调。据报道,Kaplan-Meier生存分析显示,KAT2B表达下调与肝癌患者总体生存率降低有关[24]。此外,一项有关宫颈癌的研究发现[27],KAT2B组蛋白乙酰转移酶活性的下降会受到癌蛋白HPV16E7表达的影响。肌动蛋白α1(actinin alpha 1,ACTN1)是参与癌症转移的骨架蛋白之一[28],它可以通过影响FAK信号通路的表达和包括CCL2在内的分泌因子的含量来调节肿瘤的微环境[29]。Cao等[30]研究中证明了黄芩中的黄酮类化合物Oroxylin A抑制ACTN1表达以灭活与癌症相关的成纤维细胞,从而抑制乳腺癌转移。另有研究发现ACTN1表达是口腔鳞状细胞癌(oral squamous cell carcinoma,OSCC)预后不良的独立预测因子,敲除ACTN1可以抑制OSCC肿瘤细胞增殖和转移[31]。虽然这些基因目前尚缺乏完善的研究来证明其与CRC相关,但它们有可能是治疗CRC的潜在重要靶标。
为了阐明每个模块的生物学功能,本研究对各模块进行通路富集分析。结果显示通路主要富集于细胞周期(hsa04110)、DNA复制(hsa03030)、Wnt信号通路(hsa04310)、PI3K-AKT信号通路(hsa04151)等。其中最显著相关的是细胞周期(hsa04110)。多项研究已证实p53基因突变、细胞周期阻滞、异常细胞分裂增殖、DNA复制等与CRC的发生发展密切相关[32-35]。先前的研究表明,人类CRC的发生中与细胞周期相关的上调基因可以预测CRC患者的整体生存情况[36]。近年来,Wnt/β-catenin途径(hsa04310)已成为CRC治疗的靶点,因为大多数CRC患者至少有一个Wnt信号级联基因发生突变,如β-catenin(CTNNB1)和腺瘤性息肉病基因。据报道,汉黄芩素通过诱导G1细胞周期阻滞和下调β-连环蛋白依赖性Wnt信号通路来抑制人CRC细胞的体外增殖[37]。Wnt信号通路是CRC中重要的失调通路之一,它与肿瘤细胞增殖和抗化疗有关[38]。研究表明,PI3K-AKT信号通路(hsa04151)在肿瘤细胞的增殖、凋亡及迁移侵袭方面发挥重要作用,并与多种肿瘤的发生发展相关[39]。我们已经证明PIK3CA和AKT1的基因变异与CRC风险的显著增加相关[40]。 Slattery等[41]发现PI3K/AKT信号通路在CRC中是失调的,并且一些失调基因与miRNAs有关。Yang等[42]研究显示GLI1与PI3K/AKT/NFκB信号的联合靶向治疗可对CRC患者产生协同治疗作用,从而为CRC的治疗提供新的途径。
本研究探讨13个功能模块与KEGG通路类别之间的相互作用。结果表明功能相似的模块之间会相互影响,在疾病的发生发展中共同发挥作用。13个功能模块KEGG通路分类主要参与信号转导、信号分子与相互作用、内分泌系统、细胞生长与死亡、免疫系统、传染病、复制与修复、聚糖的生物合成与代谢等多个功能类别。模块M2、M5、M6、M8与信号转导有关,涉及的通路有Hippo信号通路、Ras信号通路、PI3K-AKT信号通路、Wnt信号通路、cAMP信号通路。据文献报道,这一系列信号通路在CRC的发生和进展中起关键作用。模块M8、M10、M11与内分泌系统有关,胃肠道粘膜散在分布众多隐匿的神经内分泌细胞,因此被认为是最大的神经内分泌器官。有研究证实,CRC的发病与2型糖尿病之间存在关联[43]。糖尿病是一种属于内分泌系统的代谢性疾病,间接揭示CRC的发生与内分泌系统息息相关。因此,可从挖掘到的模块以及相应富集通路进一步深入了解CRC与内分泌系统之间关系的机制。
综上,本研究利用转录组数据从特异性功能模块的角度分析了CRC基因网络的功能学特征,为疾病基础研究提供新思路。本次分析共挖掘了15个功能模块,87个密切相关的核心基因(如CDK1、COL1A2、MYC等)以及发现了Wnt信号通路、PI3K-AKT信号通路、细胞周期等与CRC发病有重要意义的富集通路。这些发现不仅增加了对CRC发病机制和潜在分子机制的认识,而且为肿瘤标记物的筛选和药物靶点的选择提供了理论基础。本研究也存在一些不足,新发现的相关基因和通路功能仍需要进一步的分子生物学实验和动物实验来验证。
作者声明:本文全部作者对于研究和撰写的论文出现的不端行为承担相应责任;并承诺论文中涉及的原始图片、数据资料等已按照有关规定保存,可接受核查。
学术不端:本文在初审、返修及出版前均通过中国知网(CNKI)科技期刊学术不端文献检测系统的学术不端检测。
同行评议:经同行专家双盲外审,达到刊发要求。
利益冲突:所有作者均声明不存在利益冲突。
文章版权:本文出版前已与全体作者签署了论文授权书等协议。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
计算机应用与软件(2021年10期)2021-10-15
现代临床医学(2021年4期)2021-07-31
云南医药(2021年3期)2021-07-21
天津医科大学学报(2021年1期)2021-01-26
小型微型计算机系统(2020年5期)2020-05-14
计算机与生活(2020年5期)2020-05-13
火力与指挥控制(2020年1期)2020-03-27
中国广播(2016年11期)2016-12-26
软件导刊(2016年9期)2016-11-07
