
ERK信号通路介导的EP300过表达在苯肾上腺素诱导小鼠心肌细胞肥大中的作用*
2021-06-02 11:55黄丽欣彭波辉冯玉松罗孝美吴书琪张焕婷
中国病理生理杂志 2021年5期
黄丽欣,彭波辉,彭 昌△,冯玉松,罗孝美,吴书琪,张焕婷
(遵义医科大学1附属医院儿内一科,2第一临床学院,3基础医学院生理教研室,贵州遵义563000)
病理性心肌肥厚是引起心脏衰竭和心源性猝死的重要原因。近年来病理性心肌肥厚发病率呈逐渐上升趋势,且防治措施尚不完善[1],因此寻找新的治疗手段成为当前的研究热点。本课题组前期研究证实组蛋白乙酰化修饰失衡参与了苯肾上腺素(phenylephrine,PE)所致的病理性心肌肥厚,且进一步证实组蛋白乙酰化酶抑制剂漆树酸(anacardic acid,AA)能够逆转PE所致的心肌肥厚[2],但该过程的上游信号通路并不清楚。细胞外信号调节激酶(extracellular signal-regulated kinase,ERK)是MAPKs家族中的一个主要成员[3]。研究显示,ERK通路在细胞分化、增殖、应激反应和细胞凋亡中起着重要调节作用,且该通路可通过上调组蛋白H3乙酰化水平,参与酒精暴露所致的心脏发育相关基因过表达,提示该通路可能与组蛋白乙酰化修饰失衡介导的心肌肥厚相关[4]。因此,本研究应用PE诱导小鼠心肌细胞肥大模型,探讨ERK信号通路在组蛋白乙酰化酶抑制剂AA改善PE诱导的小鼠心肌细胞肥大中的作用,为心肌肥厚的防治提供参考资料。
材料和方法
1 实验动物
选取SPF级新生3 d的健康昆明小鼠120只,体重2.3~2.7 g,雌雄不限,由重庆医科大学动物中心提供,动物许可证号为SYXK(渝)2012-0015。
2 主要试剂与仪器
ERK抑制剂U0126、胶原酶Ⅱ和PE(Sigma);胎牛血清(Gibco);抗ERK及p-ERK单克隆抗体(Cell Signaling Technology);抗第9位赖氨酸乙酰化的组蛋白H3(acetylated histone H3 at lysine 9,H3K9ac)、β-肌球蛋白重链(β-myosin heavy chain,β-MHC)和E1A结 合 蛋 白p300(E1A-binding protein p300,EP300)单克隆抗体及兔IgG多克隆抗体(Abcam);抗Alexa Fluor 488荧光标记的山羊抗小鼠IgG荧光Ⅱ抗(中杉金桥生物技术有限公司);抗α-actin及HRP标记的Ⅱ抗(Proteintech);Trizol总RNA提取试剂(BioTeke);SDS-PAGE凝胶试剂盒(Beyotime);免疫共沉试剂盒(Beaver)。
3 主要实验方法
3.1 心肌细胞原代培养选取出生3 d的昆明小鼠,在超净工作台上用无菌镊子及眼科剪取出心脏,轻柔挤出心脏中的血,预冷PBS液洗净残血,将其剪成1 mm×1 mm×1 mm大小的碎块,加入少许0.05%胶原酶Ⅱ反复吹打消化2 min,放置沉淀后,弃上清液,如此反复,直至消化完全。将收集的上清液离心(240×g、10 min),弃上清,沉淀与4 mL含20%胎牛血清的DMEM培养液重悬后接种到25 mL培养瓶中,在培养箱(37℃、5%CO2)中差速培养90min后,将细胞悬液转移至新的培养瓶中,加入终浓度为0.1 mmol/L的BrdU后继续培养。
3.2 实验分组及细胞干预各组药物浓度参照文献[5-7]选取,采用PE干预心肌细胞48 h构建心肌细胞肥大模型后,按随机数字表法将心肌细胞分为正常(normal)组(不给予任何处理)、生理盐水(normal saline,NS)组(给予等量NS处理细胞)和PE组(用100 μmol/L PE干预心肌细胞48 h),探讨PE干预的小鼠心肌细胞中EP300和H3K9ac的表达水平及其相互调控关系。再将心肌细胞分为NS组、溶剂对照(DMSO+PE)组(用100μmol/L PE加等体积的DMSO处理心肌细胞)、PE组、AA+PE组(用50μmol/L AA处理后加入100μmol/L PE处理心肌细胞)、U0126+PE组(用10μmol/L U0126干预后加入100μmol/L PE处理心肌细胞)和AA+U0126+PE组(用50μmol/L AA孵育心肌细胞30 min后,再加入10μmol/L U0126处理,最终用100μmol/L PE干预心肌细胞48 h),进一步探讨ERK信号通路介导的EP300过表达在PE诱导小鼠心肌细胞肥大中的作用。
3.3 Western blot检测提取心肌细胞蛋白,用BCA法检测蛋白浓度,SDS-PAGE分离蛋白,转膜,将PVDF膜浸泡于封闭液中,分别加入兔来源单克隆抗体H3K9ac、EP300、β-MHC、ERK和p-ERK,4℃摇床孵育过夜,TBST洗涤,每次5 min,洗涤6次,然后加入Ⅱ抗孵育,摇床上孵育1 h,TBST洗涤,每次5 min,洗涤6次,使用化学发光试剂发光显影。采用Bio-Rad图像分析仪进行图像扫描;应用Quantity One 4.4软件进行分析。
3.4 RT-qPCR检测引物用Primer Premier 5.0软件设计由宝生物公司合成。将β-MHC产物进行梯度稀释,运用Bio-Rad CFX96荧光定量PCR仪扩增,做出标准曲线,得到R2值和扩增效率。β-MHC的上游引物序列为5′-CAAAGGCAAGGCAAAGAAAG-3′,下游引物序列为5′-GCAGAGGCACCAAAATGAAT-3′,产物大小为113 bp;β-actin的上游引物序列为5′-CCTTTATCGGTATGGAGTCTGCG-3′,下游引物序列5′-CCTGACATGACGTTGTTGGCA-3′,产 物 大 小 为289 bp。反应条件为:94℃30 s;94℃5 s,60℃30 s,40个循环。以β-actin作为内参照,所得数据用PCR仪自带的基于Pfaffl原理的相对定量数据分析软件处理。
3.5 免疫共沉淀移去培养液,PBS清洗,用结合缓冲液(含蛋白酶抑制剂)裂解细胞,充分混匀后置于冰上孵育10 min,低温(4℃)20 000×g离心10 min,收集上清。将磁珠预处理后,分别加入EP300抗体和H3K9ac抗体,室温翻转20 min,弃上清,加入缓冲液洗涤2次后,加入抗原4℃翻转孵育过夜。将抗原吸附后的磁体/抗体/抗原复合物经磁性分离,再洗涤,弃掉上清,加入5×SDS-PAGELoading Buffer混合均匀,95℃加热5 min,室温下18 000×g离心10 min,收集上清液进行Western blot检测。
3.6 免疫荧光检测制作细胞爬片,吸出细胞培养液,PBS洗涤,4%多聚甲醛固定20 min,PBS洗涤,每次5 min,洗涤3次,0.3%Triton X-100室温孵育15 min,PBS洗涤,每次5 min,洗涤3次,室温下山羊血清封闭30 min,加入抗α-actin(1∶50)4℃孵育过夜,PBS洗涤3次,每次5 min,Alexa Fluor 488标记的山羊抗小鼠IgGⅡ抗(1∶1 000),室温避光孵育1.5 h,PBS洗涤3次,每次5 min,再加入抗EP300Ⅰ抗4℃孵育过夜,PBS洗涤3次,每次5 min,孵育Ⅱ抗,室温避光孵育1.5 h,PBS洗涤3次,每次5 min,DAPI染核5 min,PBS洗涤3次,每次5 min,抗荧光淬灭剂封片,于荧光倒置显微镜下观察,应用Image-Pro Plus专业图像分析软件分析。
4 统计学处理
所有数据用均数±标准差(mean±SD)表示,采用SPSS 18.0统计软件进行统计学分析。多组间比较采用单因素方差分析,组间均数比较应用SNK-q检验,以P<0.05为差异有统计学意义。
结 果
1 PE干预的小鼠心肌细胞中EP300和H3K9ac的表达水平及其相互调控关系
Western blot结果显示,在培养的小鼠心肌细胞中,PE组EP300和H3K9ac表达水平显著高于NS组(P<0.05),见图1A、B。免疫共沉淀结果显示,EP300可特异性与H3K9ac相互结合,见图1C。
2 各组小鼠心肌细胞中ERK和p-ERK的表达水平
Western blot结果显示,在培养的小鼠心肌细胞中,ERK表达水平在各组心肌细胞中无显著差异(P>0.05),而p-ERK的表达水平在PE组显著高于生理盐水对照组(P<0.05);而ERK抑制剂和AA干预组的p-ERK水平均显著低于PE组(P<0.05),见图2。
3 各组小鼠心肌细胞中H3K9ac水平
Western blot结果显示,在小鼠心肌细胞中,PE组H3K9ac水平显著高于NS组(P<0.05);而ERK抑制剂和AA干预组H3K9ac的水平均显著低于PE组(P<0.05),见图3。
4 各组小鼠心肌细胞中组蛋白乙酰化酶EP300的表达水平
免疫荧光及Western blot结果表明,在培养的小鼠心肌细胞中,PE组EP300表达水平显著高于NS组(P<0.05);AA及ERK抑制剂干预组EP300的表达水平显著低于PE组(P<0.05),见图4。
5 ERK信号通路在AA逆转PE诱导心肌肥大标志物β-MHC过表达中的作用
RT-qPCR及Western blot结果显示,PE组心肌肥大标志物β-MHC的mRNA水平及蛋白水平均显著高于NS组(P<0.05),ERK抑制剂及AA干预组β-MHC的mRNA及蛋白水平均显著低于PE组(P<0.05),见图5。
讨 论
心肌肥厚是心肌细胞对各种刺激因素所产生的适应性反应,分为生理性肥厚和病理性肥厚,病理性心肌肥厚主要表现为心肌细胞体积增大和心肌间质增生,是导致慢性心力衰竭和心源性猝死的独立危险因素[8]。本课题组前期研究证实组蛋白乙酰化修饰失衡参与了PE诱导的小鼠心肌肥厚[9],而AA能够抑制组蛋白乙酰化酶(histone acetylases,HATs)介导的组蛋白H3K9高乙酰化进而改善心肌肥厚[10],且进一步证实P38丝裂原活化蛋白激酶(P38 mitogenactivated protein kinase,P38 MAPK)信号通路参与了AA改善PE诱导的心肌肥厚[11],但MAPK信号通路家族还包括c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)和ERK信号通路,而ERK和JNK信号通路是否参与AA改善PE诱导小鼠心肌肥厚并不清楚。因此,本研究应用PE诱导小鼠心肌细胞肥大,来探讨ERK信号通路在EP300介导的组蛋白H3K9ac乙酰化失衡致心肌细胞肥大中的作用。
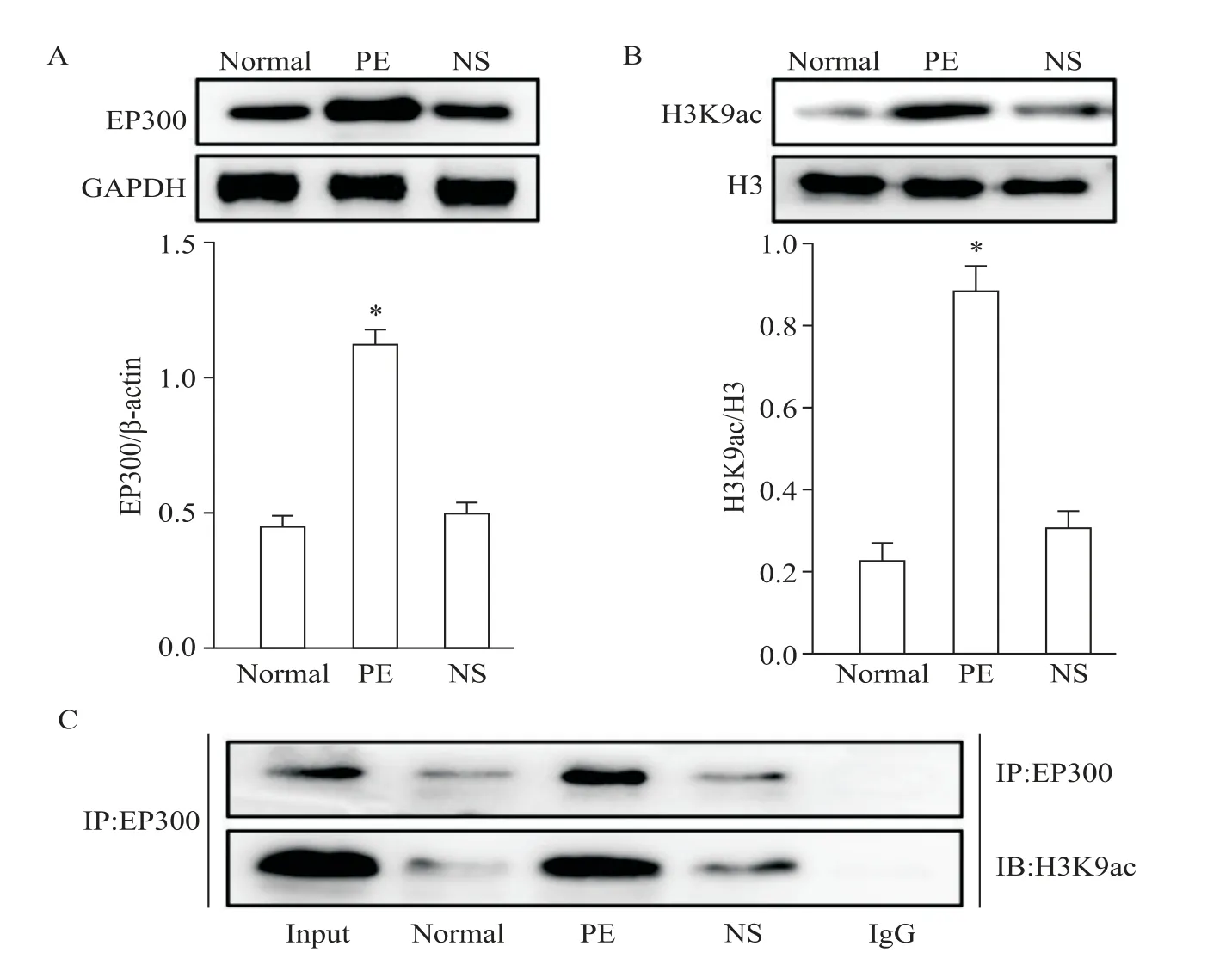
Figure 1.The expression of EP300 and H3K9ac in the mouse myocardial cells and their mutual regulation.A:the protein levels of EP300;B:the protein levels of H3K9ac;C:co-immunoprecipitation(Co-IP)in cell lysates of mouse myocardial cells exposed to three different experimental conditionswith anti-EP300-protein Gmagnetic beads and immunoblot(IB)with an anti-H3K9ac or anti-EP300 antibody for evaluation of protein expression.Input:positive control;IgG:negative control.Mean±SD.n=3.*P<0.05 vs NSgroup.图1 小鼠心肌细胞中EP300和H3K 9ac的表达水平及其相互调控关系
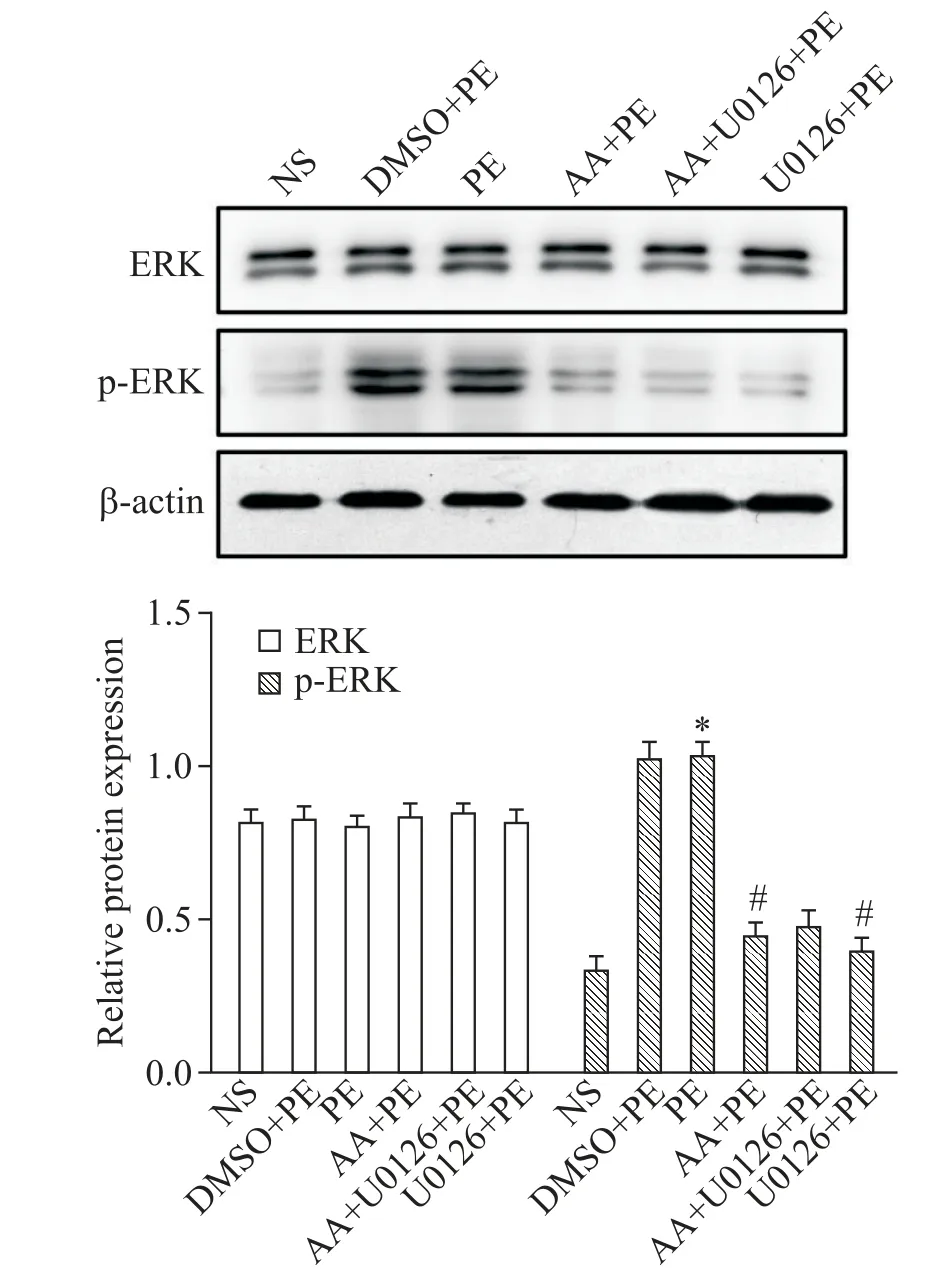
Figure 2.The protein levels of ERK and p-ERK in the mouse myocardial cells with different treatments.Mean±SD.n=6.*P<0.05 vs NSgroup;#P<0.05 vs PEgroup.图2 各组小鼠心肌细胞中ERK及p-ERK的蛋白水平
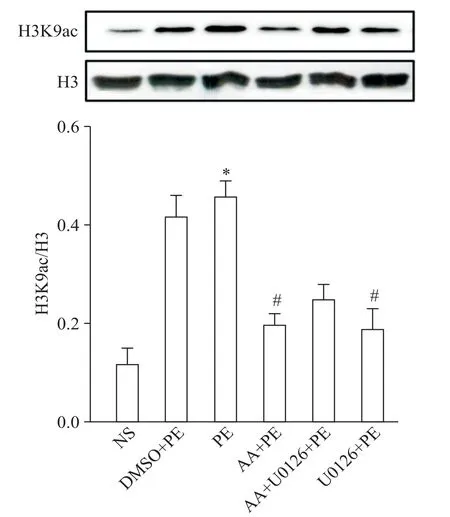
Figure 3.The level of H3K9ac in the mouse myocardial cells with different treatments.Mean±SD.n=6.*P<0.05 vs NSgroup;#P<0.05 vs PEgroup.图3 小鼠心肌细胞中组蛋白H3K9ac水平的比较
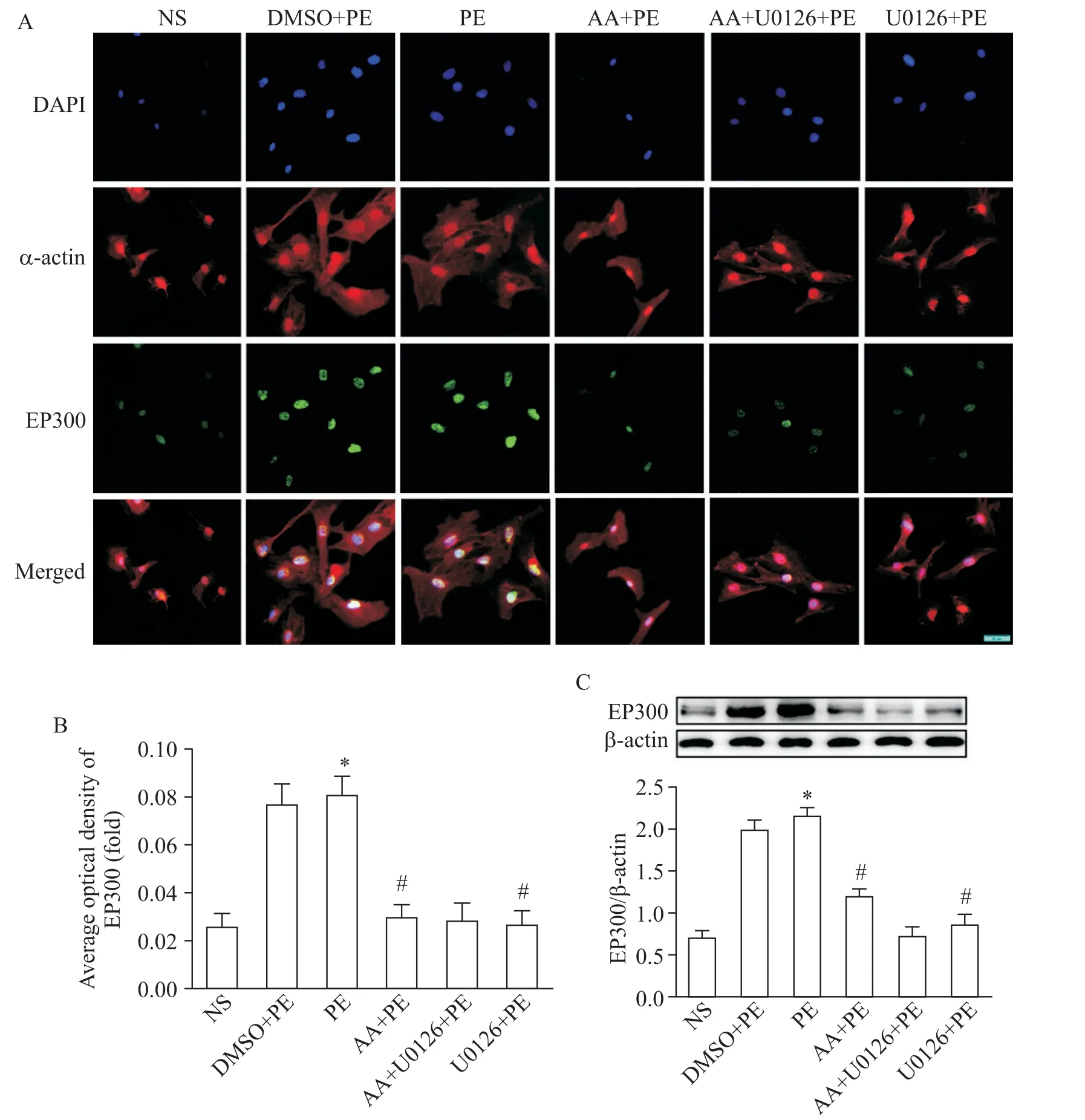
Figure 4.The expression of EP300 in the mouse myocardial cells with different treatments.A:the immunofluorescence images of myocardial cells(scale bar=20μm);B:the EP300 average optical density;C:the protein levels of EP300 were assayed by Western blot.Mean±SD.n=6.*P<0.05 vs NSgroup;#P<0.05 vs PEgroup.图4 小鼠心肌细胞中组蛋白乙酰化酶EP300的表达水平
组蛋白乙酰化与基因的活化有着密切关系,而去乙酰化与基因沉默有关,组蛋白乙酰化/去乙酰化失衡在心肌肥厚中发挥着重要作用。组蛋白乙酰化通过HATs介导,HATs在心脏表达的亚型主要包括:EP300、p300/CBP辅助因子(p300/CBP-associated factor,PCAF)、组蛋白乙酰化酶氨合成通用控制蛋白5(general control nonderepressible-5,GCN5)、CREB结合蛋白(CREB-binding protein,CBP)和类固醇受体辅活化子1(steroid receptor coactivator-1,SRC1)[12-13]。EP300能改变染色质结合转录因子,松解染色质结构而影响基因表达。EP300/CBP作为一种转录辅助激活因子,能与多种转录因子互相作用,是介导心肌肥厚的关键因子[14]。EP300和CBP共同调控心脏肥厚相关基因过表达,可能在PE诱导的心肌肥大中发挥重要作用。本研究结果显示,在PE诱导的小鼠心肌肥大细胞中,EP300及组蛋白H3K9ac水平显著高于生理盐水对照组,为了进一步证实两者的关系,我们进一步应用免疫共沉淀验证,结果表明组蛋白H3K9ac受EP300直接调控。以上结果提示EP300过表达介导的组蛋白H3K9乙酰化修饰失衡可能参与了心肌肥厚发生、发展的病理过程。但是组蛋白乙酰化酶EP300介导的其他组蛋白(如:H3K27ac)修饰是否参与了病理性心肌肥厚过程,需进一步证实。
研究显示在病理性心肌肥厚大鼠心肌细胞中,ERK磷酸化水平较高,降低其水平可改善心肌肥厚[15]。研究证实ERK在心肌细胞中的下游靶点包括EP300和CBP等[16-17],表明ERK能够调控EP300的表达。当ERK受到体内、体外刺激时,经过一系列反应发生磷酸化而激活,进一步诱导肥大相关基因表达。本研究结果证实PE组p-ERK水平显著升高,与EP300变化水平相一致,但ERK水平并没有明显变化,表明ERK并没有参与PE诱导心肌肥厚的过程,而ERK可能在上述病理过程中发挥了重要作用,同时也提示在PE诱导的心肌肥厚小鼠心肌组织中ERK信号通路处于激活状态,参与心肌肥厚的信号通路较多,其他信号通路是否在PE诱导的小鼠心肌肥厚组织中也处于激活状态(如JNK)尚待进一步研究。研究证实PE所致的心肌细胞肥大中MEF-2和β-MHC会显著升高[18],β-MHC是常用的心肌肥厚标志物,在心脏收缩功能中起着重要的作用,因此我们进一步检测了β-MHC的转录水平及蛋白水平。结果显示,PE组小鼠心肌细胞肥大标志物β-MHC mRNA水平及蛋白水平均显著升高,表明β-MHC是判断PE诱导心肌细胞肥大的重要标志物之一。为进一步证实ERK信号通路对EP300介导的组蛋白H3K9ac乙酰化失衡在PE诱导小鼠心肌细胞肥大中的作用。本实验应用ERK抑制剂U0126及组蛋白乙酰化酶抑制剂AA干预PE处理的小鼠心肌细胞,结果表明分别经过上述两种抑制剂干预后的小鼠心肌细胞中p-ERK、H3K9ac、β-MHC及EP300表达水平与单纯PE处理组相比均显著降低,进一步提示我们ERK信号通路参与的EP300介导的组蛋白乙酰化失衡参与了PE诱导的小鼠心肌细胞肥大。
综上所述,ERK信号通路在EP300介导的组蛋白H3K9ac乙酰化失衡致PE诱导小鼠心肌细胞肥大中扮演着重要的角色,提示ERK信号通路可能成为心肌肥厚防治的新靶点。
猜你喜欢
中国生物化学与分子生物学报(2022年8期)2022-09-08
世界科学技术-中医药现代化(2022年2期)2022-05-25
世界科学技术-中医药现代化(2021年7期)2021-11-04
中成药(2017年9期)2017-12-19
中成药(2017年5期)2017-06-13
海南医学(2016年8期)2016-06-08
中国组织化学与细胞化学杂志(2016年4期)2016-02-27
中国病理生理杂志(2015年8期)2015-12-21
华南农业大学学报(2015年5期)2015-12-04
中国当代医药(2015年16期)2015-03-01
