
Thioredoxin-interacting protein contributesto cardiac fibrosisby elevating oxidative stress in cardiac fibroblasts*
2021-06-02 11:55PENGLinqianWANGRuiyuLIXingbingYANGXiyangCHENGZheDingyiYANJianghongZHANGMeixiaSHANGFeifeiYANGJiadan
中国病理生理杂志 2021年5期
PENGLin-qian,WANGRui-yu,LIXing-bing,YANGXi-yang,CHENGZhe,LÜDing-yi,YANJiang-hong,ZHANGMei-xia,SHANGFei-fei,YANGJia-dan
(1 Department of Cardiology,The First Affiliated Hospital,2Instituteof Life Science,3Department of Pharmacy,The First Affiliated Hospital,Chongqing Medical University,Chongqing 400016,China.E-mail:yangjiadan11@126.com)
[ABSTRACT] AIM:To explore the role of thioredoxin-interacting protein(TXNIP)in mediating cardiac fibrosis.METHODS:In vivo,4 weeks of angiotensin(Ang)II infusion was used to induced fibrogenesis in the heart of the mice.In vitro,cardiac fibroblasts(CFs)isolated from neonatal rats were treated with Ang IIat different concentrations for different durations.The protein expression of TXNIPin the hearts of the mice and CFs was detected by Western blot.The CFs were pretreated with losartan(an Ang IItype 1 receptor blocker)prior to Ang IIstimulation,and then TXNIPexpression was detected by immunofluorescence and Western blot.Subsequently,after knock-down of TXNIP by siRNA,multiple methods including wound healing assay,crystal violet staining,Western blot and ROSdetection were used to analyze the changes in cell migration,expression of fibrosis-related proteins collagen type I(Col I),collagen type III(Col III),vimentin andα-smooth muscle actin(α-SMA),oxidative stress and MAPK signaling in CFs exposed to Ang II.RESULTS:In vivo,TXNIPwas up-regulated in the hearts of mice with cardiac fibrosis.In vitro,TXNIPwas induced by Ang IIin a dose-and time-dependent manner,but this increase was reversed by losartan.TXNIP knock-down inhibited Ang IIinduced cell migration and decreased the protein levels ofα-SMA,vimentin,Col Iand Col IIIin Ang II-treated CFs.Notably,TXNIP knock-down alleviated cellular oxidative stress and suppressed the activated MAPK signaling pathway in CFs under Ang II stimulation.CONCLUSION:TXNIP may participate in the pathogenesis of cardiac fibrosis through promoting oxidative stress via MAPK signaling pathway,suggesting that TXNIP may be a promising therapeutic target for treatment of cardiac fibrosis.
[KEY WORDS] Cardiac fibrosis;Thioredoxin-interacting protein;Oxidative stress;Angiotensin II;MAPK signaling pathway
Cardiac fibrosis is a biological process that is involved in hypertension,cardiomyopathy,coronary atherosclerosis,myocardial infarction and almost all forms of pathophysiological cardiac conditions[1].Immoderate cardiac fibrosis activation characterized by excessive proliferation of fibroblasts,myofibroblast generation and extracellular matrix(ECM)deposition often results incardiacremodeling,thereby impairing cardiac function.Indeed,oxidative stress has been implicated as an important molecular mechanism that promotes cardiac fibrosis,and antioxidant treatment exerts antifibrotic effects and partially mitigates cardiac remodeling[2].
As a crucial redox-sensitive protein,thioredoxininteracting protein(TXNIP)is sensitive to pathological stimuli and participates in a variety of biological functions[3].Preclinical studies have shown that TXNIP expression was sharply increased in renal fibrosis and pulmonary fibrosis,and TXNIP inhibition effectively ameliorated the progression of these fibrotic diseases[4-5].Furthermore,recent studies suggested that TXNIPdeficiency improved cardiac dysfunction in mice with pressure overload and myocardial ischemia/reperfusion[6-7],indicating that TXNIP may be a detrimental factor in cardiac disorders.However,little is currently known about the role of TXNIPin cardiac fibrosis.
The renin-angiotensin system(RAS)is closely linked to the pathogenesis of cardiac fibrosis[8].As a hallmark of RAS activation,angiotensin(Ang)II is widely used to activate cardiac fibrosis bothin vivoandin vitro[9].Therefore,in the present study,a murine model of Ang II infusion and Ang II-treated cardiac fibroblasts(CFs)were used to determine the alterations in TXNIP expression.We also explored the potential regulatory effects of TXNIPon cardiac fibrogenesis.
MATERIALSAND METHODS
1 Animals and experimental protocols
Male C57/BL6 mice(8 weeks old,n=20)were obtained from Experimental Animal Centre of Chongqing Medical University(Chongqing,China).The protocols for animal experiments were performed in accordance with the guidelines of the Ethics Committee of Chongqing Medical University and in compliance with the Guidelines for the Care and Use of Laboratory Animals published by the National Institutes of Health(Bethesda,MD,USA).All mice were housed with free access to standard mouse chow and water.
Firstly,the mice were randomly divided into saline and Ang II infusion groups(n=10 per group).Then,the mice were anaesthetized with an intraperitoneal injection of pentobarbitone sodium(Merck,Germany)at a dose of 50 mg/kg,and an osmotic minipump(Alza Corp,CA,USA)was subcutaneously implanted in the interscapular space of each mouse.Whereafter,Ang II(1 000 ng·kg-1·min-1;Sigma,MO,USA)or an equal volume of saline was steadily released at a flow of 0.25μL/h.At 4 weeks after infusion,all mice were sacrificed by an overdose of pentobarbital sodium(150 mg/kg),and the hearts were then harvested for subsequent experiments.
2 Histological analysis
The hearts were fixed in 4%neutral buffered paraformaldehyde,embedded in paraffin,and sectioned at a thickness of 5μm for Masson’s trichrome and Sirius red staining(all from Solarbio,Beijing,China).The fibrotic area is expressed as a percentage of the fibrotic area to the total myocardial area(exclusive of the perivascular collagen and luminal areas)and was quantitatively calculated in each specimen with Image-Pro Plus software(Media Cybernetics,MD,USA).
3 Isolation and identification of neonatal rat cardiac fibroblasts
Primary rat CFs were isolated from neonatal rats(within 7 days of birth)as previously described[10].Briefly,the hearts were rapidly excised and cut into pieces,and 0.1% collagenase type II(Mengbio,Chongqing,China)and 0.08%trypsin(Beyotime,Shanghai,China)were used to digest the heart tissues at 37℃for 30 min.After centrifugation,the cells were resuspended in complete medium containing 10%fetal bovine serum(FBS)and cultured in a humidified incubator at 37℃with 5%CO2.The cells were purified by pre-plating for 1 h;then,the non-adherent cells(mostly cardiomyocytes)in the culture medium were removed,and the adherent CFs were supplemented with new medium and sequentially cultured under standard conditions.The dynamic growth of CFs at different time points is shown in Figure 1A. Immunofluorescent staining of vimentin(a marker of fibroblasts)was used to determine the purity of the CFs(Figure 1B).The cells at the first passage were used for experiments.
4 Cell transfection
Three different small interfering RNA(siRNA)sequences targeting TXNIP and a negative control(NC)scrambled siRNA were designed and constructed by Hanbio(Shanghai,China).CFs at 30%confluence were transfected with siRNA oligos(50 nmol/L)with Lipofectamine 2000 reagent(Invitrogen,CA,USA)according to the manufacturer’s instructions.After transfection for 48 h,the CFs were harvested to determine the transfection efficiency.CFs transfected with the 3rd sequence(#3)of TXNIP-siRNA exhibited the most significant decrease in TXNIP expression(0.66-fold decrease)compared with that of the NC siRNA group(Figure 2).Therefore,TXNIP-siRNA#3 was used in the subsequent experiments.The detailed siRNA sequence targeting TXNIP was 5′-GGACGUGAUUCCUGAAGAUTT-3′,and the NC sequence was 5′-UUCUCCGAACGUGUCACGUTT-3′.
5 Immunofluorescence staining and observation
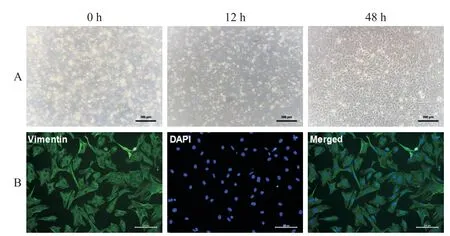
Figure 1.Isolation and identification of neonatal rat cardiac fibroblasts.Neonatal rat cardiac fibroblasts were isolated from neonatal rats(within 7 d of birth)by enzymatic digestion,and the cells at the first passage were harvested for fibroblast identification by staining for fibroblast marker vimentin.A:dynamic observation of cardiac fibroblasts at different time points under a light microscope(×200);B:immunofluorescence analysis of vimentin(green)in thecardiac fibroblasts(×100).
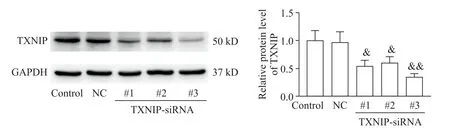
Figure 2.siRNA transfection efficiency in cardiac fibroblasts.The cardiac fibroblasts were transfected with 3 different siRNA sequences targeting TXNIP or a scrambled NCsiRNA(50 nmol/L)for 48 h,and the protein level of TXNIPwas detected by Western blot.Mean±SD.n=3.&P<0.05,&&P<0.01 vs NCgroup.
CFs were cultured on glass coverslips prior to treatment and fixed with 4%paraformaldehyde for 10 min.Then,the cells were permeabilized with 0.25%Triton X-100 and blocked with 1%bovine serum albumin for 30 min at room temperature.Subsequently,the cells were incubated with primary antibodies against TXNIP(1∶100;Proteintech)andα-smooth muscle actin(α-SMA;1∶200;Abcam)overnight at 4℃.FITC-and Cy3-labelled secondary antibodies(Beyotime,Shanghai,China)were used to label TXNIP(in green)and α-SMA(in red),respectively.DAPI was used for nuclear staining.Fluorescence signals were captured under a fluorescence microscope(Leica)and quantitatively analyzed using Image-Pro Plus software.
6 Detection of intracellular reactive oxygen species(ROS)
Intracellular ROS levels were measured with a 2′7′-dichlorodihydrofluorescein diacetate(DCFH-DA)solution(Beyotime,Shanghai,China)according to the manufacturer′s instructions.Image-Pro Plus software was used to quantitatively measure the intracellular fluorescence,which was indicative of the ROSlevel.
7 Determining the protein levels by Western blot
Proteins were extracted from mouse hearts and CFs by lysis in ice-cold RIPA lysis buffer.The protein concentration was determined using a BCA protein assay kit(Beyotime,Shanghai,China).Protein samples were separated by SDS-polyacrylamide gel electrophoresis and transferred to PVDF membranes.After being blocked with 5%non-fat milk at room temperature for 1.5 h,the membranes were incubated with primary antibodies againstα-SMA(1∶3 000;Abcam),collagen type I(Col I;1∶2 000;Proteintech),collagen type III(Col III;1∶1 000;Proteintech),vimentin(1∶2 000;Proteintech),thioredoxin(TRX;1∶1 000;Proteintech),NADPH oxidase 2(NOX2;1:1000;Santa Cruz Biotechnology),superoxide dismutase 2(SOD2;1∶2 000;Santa Cruz Biotechnology),TXNIP(1∶1 000;Proteintech),p38(1∶1 000;Bimake),p-p38(Thr180/Tyr182;1∶1 000;Immunoway),JNK(1∶2 000;Proteintech),p-JNK(Thr183/Tyr221;1∶1 000;Abcam),ERK(1:1 000;Cell Signaling Technology),p-ERK(Thr202/Tyr204;1∶1 000;Cell Signaling Technology),and GAPDH(1∶5 000;Proteintech)overnight at 4℃.Then,the membranes were incubated with HRP-conjugated secondary antibodies(1∶5 000,Proteintech)for 1.5 h at room temperature.The protein bands were visualized by an enhanced ECL kit(Beyotime,Shanghai,China).GAPDH served as an endogenous control for equal sample loading.
8 Wound healing assay
Transfected CFs were incubated until 100%confluence in 6-well plates and then wounded with a 200 μL pipette tip.The cells were washed twice with PBS to remove cell debris and then cultured with or without Ang II at 10-6mol/L for 12 h.Wound closure was observed and photographed at 0 h and 12 h.The migration distance was quantified and calculated according to the following formula:[(distance at 0 h-distance at 12 h after scratching)/distance at 0 h]×100%.
9 Transwell assay
The Transwell migration assay was performed in 24-well plates with Transwell chambers(8μm;Biofil,Guangzhou,China).After transfection for 24 h,10 000 cells were seeded in the upper chambers with serum-free medium,and complete growth medium with or without Ang II(10-6mol/L)was added to the lower chambers.After treatment for 12 h,the CFs on the upper side of the filters were removed with a cotton swab and washed with PBS.Then,the cells on the lower side of the filters were stained with 1%crystal violet(Beyotime)for 30 min at room temperature.The cells on the lower side of the filters were considered as the migrated cells,and the migrated cell number of each filter was counted using Image-Pro Plus software.
10 Statistical analysis
Statistical analyses were performed using SPSS 20.0 software.All data were presented as mean±standard deviation(SD).One-way ANOVA followed by Tukey′s post hoc test was used to assess statistical significance among groups.A value ofP<0.05 was considered statistically significant.
RESULTS
1 TXNIP expression was up-regulated during cardiac fibrosis
In vivo,both Masson trichrome staining and Sirius red staining showed significant collagen deposition in the hearts of the mice after 4 weeks of Ang II infusion.Quantitative analysis showed that the fibrotic area in Ang IIgroup was obviously increased compared with saline group[(10.32±0.79)%vs(2.58±0.41)%,P<0.01,Figure 3A].In addition,the results of Western blot showed that the protein levels of Col I and Col III in Ang II group were considerably higher than those in saline group(bothP<0.01,Figure 3B),suggesting that the animal model of cardiac fibrosis was successfully established.Moreover,TXNIP in the heart tissues was markedly up-regulated in the progression of cardiac fibrosis(P<0.01,Figure 3B).
In vitro,as the concentration of Ang II increased(from 10-8to 10-5mol/L),the expression of TXNIP in the CFs was gradually up-regulated as compared with control group(Figure 3C).In addition,with the prolongation of Ang II treatment(from 6 to 48 h),TXNIP also showed an upward trend and peaked at 24 h(Figure 3C).Subsequently,we found that pretreatment with losartan(an angiotensin II type 1 receptor blocker)eliminated the increase in TXNIP expression(Figure 3D).Furthermore,immunofluorescence analysis showed that Ang II induced sharp increases in TXNIPexpression both in the nucleus and cytoplasm of CFs,and such increases were reversed by losartan pretreatment(P<0.01,Figure 3E).
2 TXNIP knock-down suppressed Ang II-induced migration of CFs
In the wound healing assay,Ang IIstimulation obviously increased the migration distance of CFs(Figure 4A).As compared with Ang II+NC-siRNA group,the migration distance of CFs was considerably reduced in Ang II+TXNIP-siRNA group(P<0.05,Figure 4A).Furthermore,as shown in Figure 4B,only minimal migrated cells in control group were observed.However,after Ang II treatment,the number of migrated cells was markedly increased(P<0.01,Figure 4B).Knockdown ofTXNIPsignificantly reduced the level of such increase(P<0.05,Figure 4B).
3 Knock-down of TXNIP alleviated Ang II-induced cardiac fibrosis activation
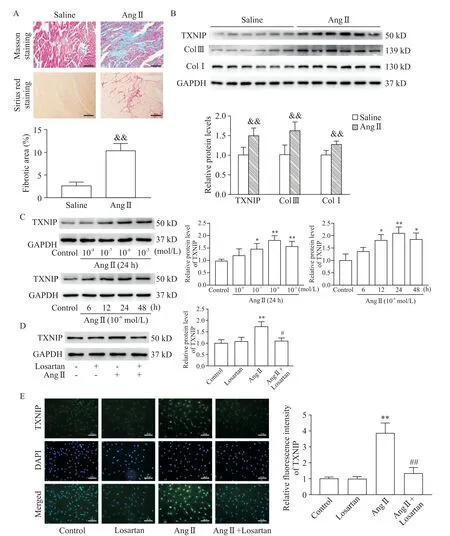
Figure 3.TXNIP expression was up-regulated during cardiac fibrosis.Mice were infused with either saline or Ang II for 4 weeks,and the hearts were harvested for histological and Western blot analysis.A:Masson staining(upper panel)and Sirius red staining(lower panel)of the hearts(×100),and quantitative analysis of the fibrotic areas in each group(n=4);B:Western blot for determining the protein levels of TXNIP,Col IIIand Col Iin the hearts(n=6);C:Western blot for determining the protein levels of TXNIPin cardiac fibroblasts treated with different concentrations of Ang IIfor different durations(n=3);D:Western blot for determining the protein levels of TXNIPin Ang II-treated cardiac fibroblasts with or without losartan pretreatment(n=3);E:cell immunofluorescence analysis of the expression and distribution of TXNIP(in green)in CFs(×200)and quantitative analysis of the relative fluorescence intensity of TXNIP in each group(n=3).Mean±SD.&&P<0.01 vs saline group;*P<0.05,**P<0.01 vs control group;#P<0.05,##P<0.01 vs Ang IIgroup.
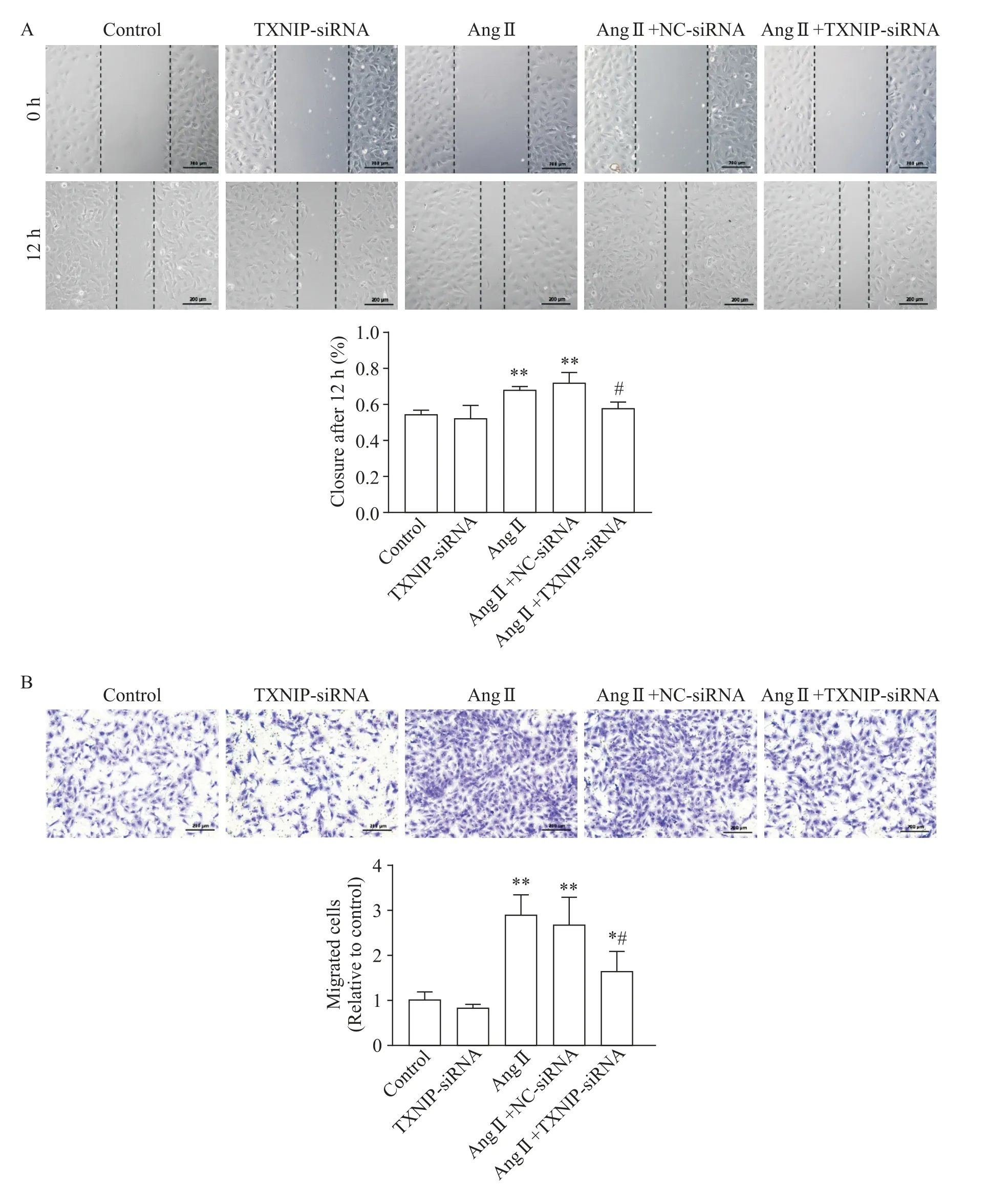
Figure 4.TXNIP knock-down suppressed Ang II-induced migration of cardiac fibroblasts.The cardiac fibroblasts were transfected with TXNIP-or NC-siRNA 24 h prior to Ang II(10-6 mol/L)stimulation.A:the scratch wound was performed at the initial time of Ang II administration,and the cells were photographed at 0 h and 12 h after wounding(×100);B:Transwell migration assay was also conducted 12 h after Ang IItreatment(×100).Mean±SD.n=3.*P<0.05,**P<0.01 vs control group;#P<0.05 vs Ang II+NC-siRNA group.
As shown in Figure 5A,the protein levels of Col III,Col I and vimentin were increased in Ang II group as compared with control group(P<0.01),while knock-down ofTXNIPeffectively mitigated the abnormities in these proteins.The expression and distribution ofα-SMA(as a marker of myofibroblast generation)were measured.Western blot analysis showed that Ang IIstimulation significantly up-regulated the expression ofα-SMA,while this up-regulation was abolished byTXNIPgene silencing(P<0.05,Figure 5A).Consistent with the protein expression data,much lower fluorescence intensity forα-SMA was also observed in Ang II+TXNIP-siRNA group than that in Ang II+NC-siRNA group(P<0.01,Figure 5B).
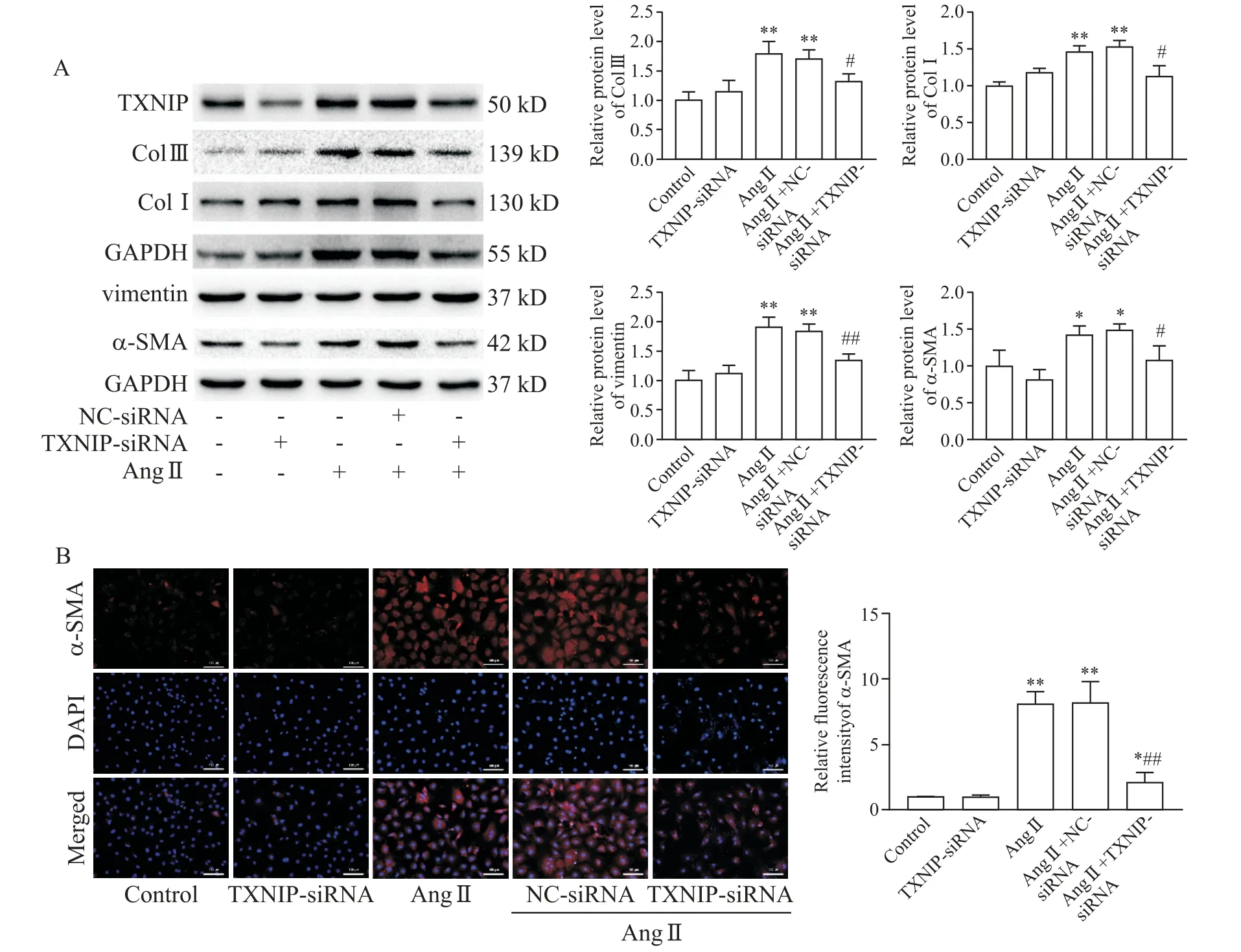
Figure 5.TXNIP knock-down alleviated Ang II-induced cardiac fibrosis activation.The cardiac fibroblasts were transfected with TXNIP-or NC-siRNA(50 nmol/L)24 h prior to Ang IIstimulation(10-6 mol/L for 24 h).A:Western blot for determining the protein levels of Col III,Col I,vimentin andα-SMA in the cardiac fibroblasts;B:immunofluorescence for analyzing the expression ofα-SMA(in red)in the cardiac fibroblasts(×200).Mean±SD.n=3.*P<0.05,**P<0.01 vs control group;#P<0.05,##P<0.01 vs Ang II+NC-siRNA group.
4 TXNIP mediated oxidative stress and promoted MAPK signaling pathway activation during cardiac fibrosis
Enhanced oxidative stress is a crucial contributing factor in the pathogenesis of cardiac fibroblasts.As shown in Figure 6A,intracellular ROSgeneration was significantly increased in Ang II group as compared with control group(P<0.01),whereas knock-down ofTXNIPmarkedly suppressed ROSgeneration in Ang IItreated CFs(P<0.01).In addition,the results of Western blot showed that the increase in NOX2 expression and decrease in SOD2 expression induced by Ang II were blunted byTXNIPsilencing(P<0.05,Figure 6B).Interestingly,as a crucial antioxidant protein,TRX expression was decreased in Ang II group as compared with control group(P<0.01,Figure 6B),while it was partially restored in Ang II+TXNIP-siRNA group(P<0.05).In addition,Tempol(an ROS scavenger)effectively normalized the up-regulated expression of Col III,Col I,vimentin andα-SMA in Ang II-treated CFs(Figure 6C),which exhibited a similar antifibrotic effect as that of TXNIPinhibition.
As a downstream signaling pathway regulated by TXNIP,MAPK signaling pathway plays a crucial role in the regulation of myofibrogenesis[11].To investigate the mechanism underlying the regulatory effect of TXNIPon cardiac fibrosis,p38,JNK and ERK,which are involved in the MAPK signaling pathway,were detected by Western blot.As shown in Figure 6D,under Ang II stimulation,the MAPK signaling pathway was obviously activated,as evidenced by the increased ratios of p-p38/p38,p-ERK/ERK and p-JNK/JNK in Ang II group(P<0.05).Compared with Ang II+NC-siRNA group,TXNIPknock-down considerably reduced the activation of these kinases. Therefore,inhibiting TXNIP partially blocked activation of MAPK signaling pathway in Ang II-treated CFs.
DISCUSSION
Fibrogenesis is a compensatory response to detrimental stimuli and acts as a scarring event to repair the injured myocardium.However,immoderate fibrogenesis in cardiac tissues increases the stiffness of the left ventricle and disturbs normal myocardial compliance,ultimately leading to the development of cardiac dysfunction[1].Multiple pathological factors,such as hypoxia,ischemia,and inflammatory cytokine release,trigger the fibrotic response in cardiac tissues[12].In addition to directly activating the profibrotic signaling pathway,these factors often lead to oxidative stress in cardiac tissues[2].The mechanistic links between oxidative stress and cardiac fibrosis have been sufficiently clarified both in preclinical and clinical practice[2,13].
As a sign of hypertrophic remodeling in the heart,fibrosis is closely related to Ang II stimulation.Therefore,Ang II is widely used to establish animal models of cardiac fibrosis and mimic fibrosisin vitrodue to its well-established profibrotic effect[9]. Although the mechanism underlying the pathogenesis of Ang II-induced cardiac fibrosis is multifactorial,oxidative stress is an indispensable factor[14].Evidence has emphasized that Ang IIincreases NADPH-dependent superoxide anion production and impairs mitochondrial function through binding to AT1R,which then leads to massive intracellular ROS accumulation[15].ROS plays an important role in the activation of multiple fibrosis-related signaling pathways,including TGF-β/Smad,NF-κB,MAPK,etc.[16].Of note,abundant evidence has confirmed that antioxidant therapy effectively inhibited Ang II-induced cardiac remodeling[17-18].
As a multifunctional protein,TXNIP plays a crucial role in the regulation of cellular oxidative stress,apoptosis,and other biological processes[3].Previous studies showed that TXNIP was specifically activated by hyperglycemia and inflammation,and TXNIPparticipated in the progression of diabetes,coronary artery disease,and several cerebrovascular disorders[19].Once triggered by pathological stimuli,TXNIP was markedly activated and subsequently released from the nucleus and shuttled into the cytoplasm to suppress the activity of TRX(a major antioxidant protein),thereby impairing the capability of ROS scavenges[3].In addition,TXNIP may directly augment ROSproduction by activating NADPH oxidases[20].Therefore,TXNIP may be a crucial mediator for ROS accumulation.Ang II has been reported to upregulate TXNIP via carbohydrate response element binding protein(ChREBP),which is an important transcription factor that facilitates TXNIPexpression[21].Here,we also showed that,Ang II accelerated intracellular ROSgeneration and disrupted the balance between oxidation and antioxidation in CFs.However,withTXNIPknock-down,these abnormal redox signals were effectively ameliorated.Notably,the decreased expression of TRX in Ang II-treated CFs was obviously restored afterTXNIPknock-down,indicating that the TXNIP inhibition-mediated alleviation of oxidative stress in CFs may be related to the activation of TRX.
In fact,increasing evidence has shown that TXNIP is correlated with cardiac function.Yoshioka J et al[6].reported that cardiomyocyte-specific TXNIPKOmice exhibited less cardiac hypertrophy,and better left ventricular function than wild-type mice in response to pressure overload and inhibition of TXNIP also prevented ischemia-reperfusion-induced myocardial injury[7].Our results further showed that TXNIP up-regulation was positively correlated with fibroblast activation and may serve as a crucial mediator to promote the development of cardiac fibrosis.
The MAPK signaling pathway is involved in the pathogenic processes of cardiac fibrosis[11].A previous study indicated that 3 major MAPK pathway molecules p38,ERK and JNK were activated in the cardiac tissues of mice with cardiac remodeling[22].In addition,prominent cardiac fibrosis accompanied by increased diastolic chamber stiffness was observed in transgenic mice with cardiac-specific over-expression of the p38[23].In contrast,inactivation of MAPK suppressed the development of cardiac fibrosis[24].It is well recognized that Ang II promoted the activation of MAPK signaling in various hypertrophic and fibrotic disorders[25].Consistent with this finding,our study showed that MAPK signaling was activated by Ang II in CFs,while it was obviously blunted byTXNIPknock-down,suggesting that TXNIP may be an important upstream protein responsible for activating the MAPK signaling pathway in cardiac fibrosis.
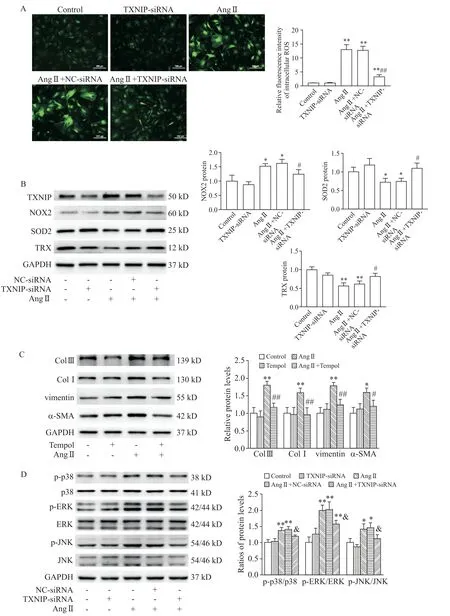
Figure 6.TXNIP mediated oxidative stress and promoted MAPK signaling pathway activation during cardiac fibrosis.The cardiac fibroblasts(CFs)were pretreated with Tempol(1 mmol/L)2 h prior to Ang IItreatment(10-6 mol/L for 24 h).A:DCFH-DA fluorescence staining of ROSin the CFs(×100);B:Western blot for determining the protein levels of several representative redox proteins NOX2,SOD2 and TRX in the CFs under different treatment conditions;C:Western blot for determining the protein levels of Col III,Col I,vimentin andα-SMA in the CFs;D:Western blot for determining the protein levels of p-p38,p38,p-JNK,JNK,p-ERK and ERK in the CFs.Mean±SD.n=3.*P<0.05,**P<0.01 vs control group;#P<0.05,##P<0.01 vs Ang IIgroup;&P<0.05 vs Ang II+NC-siRNA group.
The intrinsic links between TXNIPand the MAPK signaling pathway have been verified in renal fibrosis,metabolic syndrome,and other disorders[5,26]. A previous study suggested that silencing TXNIP abolished Aβ-induced phosphorylation of p38 MAPK in SHSY5Y cells[27].In contrast,Li et al.[28]reported that TXNIP over-expression resulted in direct activation of MAPK pathway in SMMC7221 cells.Our study showed that TXNIPregulated the activation of the MAPK pathway in Ang II-induced CFs.However,a recent study indicated that MAPK inhibition by combined treatment with p38,JNK and ERK inhibitors normalized the upregulation of TXNIP expression in propargylglycinetreated vascular endothelial cells[29].Therefore,there may be a cross-talk between TXNIP and MAPK signaling under pathological conditions.
In conclusion,the present study demonstrates that TXNIPis involved in the progression of cardiac fibrosis and that inhibition of TXNIP effectively blunts cardiac fibrosis activation by suppressing oxidative stress via MAPK signaling pathway.These findings provide novel insights into oxidative stress and fibrogenesis,which may be beneficial to explore new target for treating cardiac fibrosis.
