
微量苯酚降解中间体LLE-HPLC检测条件优化
2021-05-20 12:12畅彤崔建国张峰
应用化工 2021年4期
畅彤,崔建国,张峰
(1.太原理工大学 环境科学与工程学院,山西 晋中 030600;2.山西省市政工程研究生教育创新中心,山西 晋中 030600)
酚类微污染水应急处理是一种亟待解决的环境与健康问题[1]。微污染水处理过程中,苯酚降解产生的主要中间体对苯二酚和对苯醌的毒性危害是关注的焦点[2]。
目前,有机微污染原水采用高锰酸钾+活性炭工艺处理[3],降解过程有毒中间体较多,因此研究用高铁酸钾替代[4-5]。进厂原水中挥发酚(以苯酚计)的浓度应低于2 μg/L[6]。污染物的低浓度和高铁酸钾降解苯酚的高效性[7],使得残留污染浓度很低,现有检测方法很难达到检测要求[8-9]。
液液萃取-高效液相色谱联用(LLE-HPLC)操作简便,较为经济[10],其中液液盐析简单快速、高倍富集[11-12],但局限较大[13],必须优化条件提高精度。本文以微量苯酚及对苯二酚和对苯醌检测为例,研究优化条件,以期检测精度双优。
1 实验部分
1.1 试剂与仪器
无水苯酚、对苯二酚、对苯醌、高铁酸钾、氨水(25%)、硫酸、无水硫酸钠、硫酸铵、乙酸乙酯、冰乙酸等均为分析纯;乙腈、甲醇为色谱纯,由Millipore Milli-Q超纯水系统配制所需溶液。
LC-100 液相色谱仪;色谱柱(5 μm×4.6 mm×250 mm);DHG-9070B电热恒温鼓风干燥箱;FE28 pH计;LD-4台式离心机;50 mL离心管;THZ-82B数显恒温振荡器;DCY-SY12型水浴氮吹仪;STP FA2004天平;JY 0002电子天平;1 mL微量注射器等。
1.2 标准溶液配制
1.2.1 液相色谱检测条件实验溶液 使用色谱甲醇及色谱乙腈分别配制苯酚、对苯二酚、对苯醌均为1 000 mg/L的标准储备溶液,4 ℃避光保存,当天检测。使用前用色谱级甲醇或色谱级乙腈逐级稀释到0.1 mg/L,现用现配。
1.2.2 萃取条件实验溶液 使用超纯水配制苯酚、对苯二酚、对苯醌、顺丁烯二酸、丁二酸、草酸均为1 000 mg/L 的标准储备溶液,4 ℃避光保存,当天检测。使用前,用超纯水逐级稀释到2 μg/L,现用现配。
1.3 实验方法
1.3.1 液相色谱条件 波长260 mm,流速0.8 mL/min, 柱温30 ℃,进样量20.0 μL。采用色谱峰的保留时间定性,外标法峰高定量。流动相为甲醇∶水=40∶60。
1.3.2 萃取方法 取24 mL稀释至2 μg/L的标准工作溶液放入50 mL离心管中,加入13.0 g硫酸铵,用移液枪精准量取并加入400 μL 萃取剂,振荡使硫酸铵溶解。置于振荡器中以200 r/min速率振荡萃取15 min。取出,放在离心机中,以1 500 r/min离心5 min。取出,置于试管架上,用1 mL微量注射器反复取样,直到无法观测到两相分层。将萃取溶液经过无水硫酸钠吸水后置于2 mL进样瓶中,使用后的无水乙酸钠用少量萃取剂冲洗,将冲洗液一并置于进样瓶中。若使用乙酸乙酯作为萃取剂,还应在置于进样瓶后使用小流量氮气氮吹进样瓶中液体,使乙酸乙酯浓缩到0.1 mL左右,再以1 mL色谱甲醇冲洗瓶壁,再次氮吹浓缩至液体0.1 mL后,用色谱甲醇将液体定容至1 mL。以微量注射器吸取瓶中液体检测。每组实验平行检测3次,取平均值。
2 结果与讨论
2.1 萃取条件优化
2.1.1 萃取剂选择 目前,液液萃取法提取多种酚类化合物的方法中,主要采用的萃取剂种类有二氯甲烷、乙酸乙酯、异丙醇、正辛醇等[14-15],本文考察了二氯甲烷、乙酸乙酯、异丙醇、正辛醇对苯酚及其降解产物的富集作用和回收率,结果见表1。

表1 萃取剂富集倍数
由表1可知,二氯甲烷对苯酚萃取效果较差,且回收率明显不如乙酸乙酯,同时二氯甲烷毒性大,不如乙酸乙酯安全环保。异丙醇富集倍数能达到大约75.0倍,但是对苯酚及对苯醌的富集因子较乙酸乙酯仍有差距,且对苯酚开环羧酸产物萃取效果不如乙酸乙酯。根据范云场等的研究[16],正辛醇对苯酚及对苯二酚的富集效果较为稳定。实验结果表明,正辛醇对降解产物的富集倍数不能满足实验需求。综合考虑,最终选用可以通过氮吹进一步浓缩来提高富集倍数的乙酸乙酯作为萃取剂。
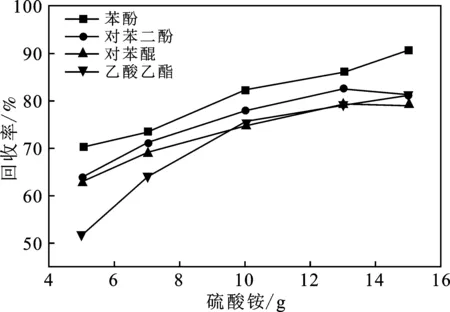
图1 硫酸铵、氯化铵质量对回收率的影响
由图1可知,硫酸铵的盐析效果优于氯化铵,故选择硫酸铵作为盐析效应中添加的盐种类。随着硫酸铵质量增加,溶剂与各物质回收率逐渐增加,其中苯酚回收率稳步上升,对苯二酚、对苯醌回收率低于苯酚,这主要是因为二者在水中溶解度大于苯酚,有部分物质未被萃取所致。但是硫酸铵继续增加时,却出现了对苯二酚和对苯醌回收率反而下降的情况,猜测是由于水中离子浓度过高反而影响到了乙酸乙酯对这2种物质在乙酸乙酯-水体系中的重分配[20]。同时乙酸乙酯回收率也在增加,这主要是因为盐溶于水,改变了水样极性,从而使乙酸乙酯更容易与水样分离[21]。实验结果表明,投加13 g硫酸铵的时候,可以得到对3种物质的最优富集效果。
2.1.3 萃取时间确定 考察了萃取时间对苯酚、对苯二酚和对苯醌3种产物及乙酸乙酯萃取剂的回收率,结果见图2。
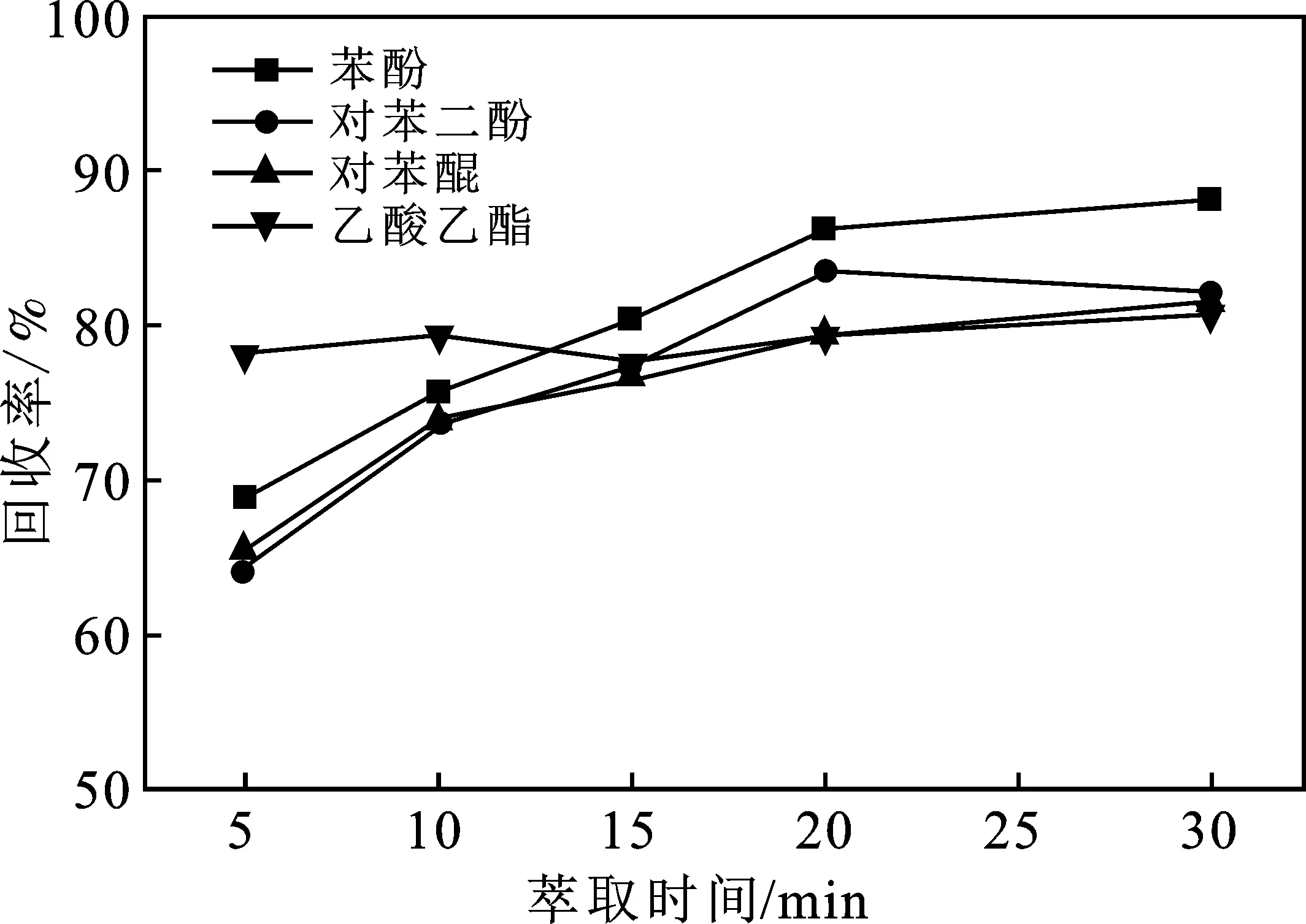
图2 萃取时间对回收率的影响
由图2可知,硫酸铵13.0 g萃取时间取20 min左右时,对苯酚等3种待测物质能基本达到最优萃取效率,时间继续增长对萃取结果影响不大。20 min 后,对苯二酚回收率下降,猜测是因为萃取过程与空气接触,产生氧化的原因。故萃取时间选择20 min。
2.2 HPLC分析条件优化
2.2.1 波长选择 经过对标准储备溶液的最大吸收波长扫描,可知苯酚及其降解产物最大吸收波长在180~280 mm范围内。在波长为260 mm的情况下,可以使几种物质得到较好的分离,拖尾减弱,峰型良好。
2.2.2 流动相选择 一些测定苯酚降解产物的实验中采用乙腈-水体系作为流动相[22-23]。实验结果表明,在同样物质浓度的情况下,使用乙腈-水流动相体系虽然在物质保留时间和拖尾因子上比甲醇-水流动相体系更有优势,但是考虑到出峰总用时及待测物质响应值大小程度,甲醇-水体系无疑是更优选择。在实际情况下能够更好体现几种有毒降解中间体。
在一些多种苯系物同时HPLC测定的情况下,在采用甲醇-水流动相体系的同时在流动相纯水组分中添加1%的冰乙酸或甲酸[24-25]。因为苯酚、苯二酚的酚羟基会在纯水中电离,从而在C18色谱柱上造成色谱峰拖尾的现象[26]。同时需要注意的是,因为实验中采用的是纯甲醇作为溶剂,出峰中还存在溶剂峰,所以在出峰情况不佳的情况下很难判断物质峰归属。实验发现,添加冰乙酸虽然利于部分待测物以分子形式存在,抑制酚类电离,但是对苯酚降解开环后羧酸产物的峰型及峰值存在一定影响,在保留时间接近的情况下反而会出现羧酸产物拖尾盖住对苯二酚的情况,实际能测出的物质峰只有3个,故不选择。由此可知,选用甲醇-水体系作为流动相是比较合适的。
2.2.3 流动相优化 实验中发现,当流动相中甲醇组分占比升高的时候,待测化合物的保留时间不断缩短,同时拖尾因子也比较小,峰型对称,出峰情况良好。但是,因为保留时间接近,导致物质与物质峰之间出现叠合,造成无法判断物质种类及其定量问题。所以,在综合考虑出峰情况、出峰总时长之后,在苯酚、对苯二酚、对苯醌均为0.1 mg/L的条件下完成了甲醇∶水不同比例的响应值实验,结果见表2。
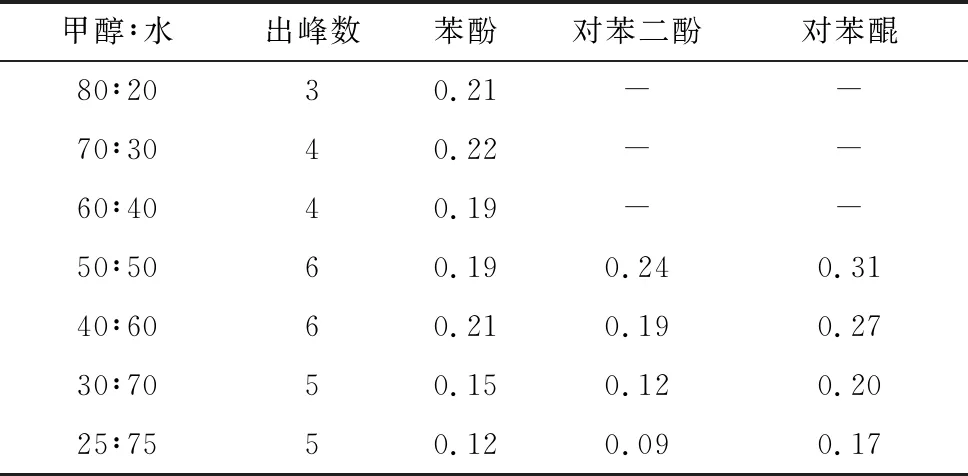
表2 甲醇/水体系流动相苯酚及中间体响应值
由表2可知,随着甲醇在流动相中组分比例减少,3种待测物质峰高均有降低,这是因为甲醇组分的减少,降低了3种物质的洗脱度,加重拖尾现象。甲醇∶水=50∶50时,由于草酸和其他物质峰过于接近,不利于对几种苯环开环后羧酸类物质的测定,考虑对草酸和顺丁烯二酸的测定需求,选择甲醇∶水=40∶60作为流动相。在流速为0.8 mL/min,波长为260 mm的条件下,可以实现对几种待测物质的有效分离,且因保留时间靠前,所需总时长较短,客观上提高了测定效率。对主要检测物质苯酚、对苯二酚和对苯醌都有较为良好的检测效果。
2.2.4 标线测定 以色谱甲醇分五级逐级稀释标准储备液至0.05~10.0 mg/L,得到苯酚、对苯二酚、对苯醌3个待测物标准溶液,以各化合物峰高作为考量依据,进行线性回归分析,结果见表3。
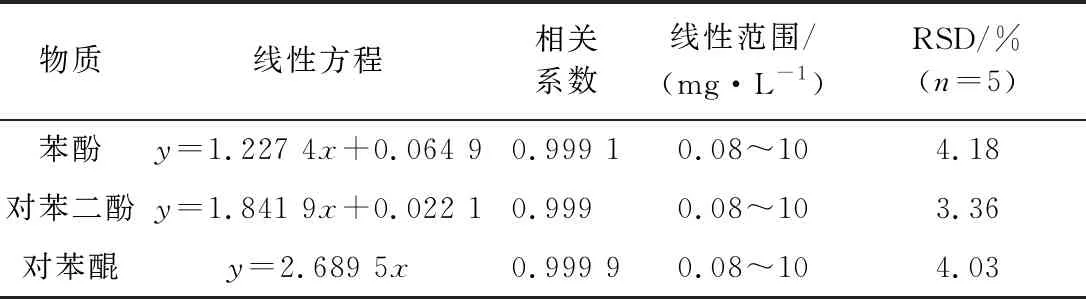
表3 化合物线性参数
2.3 检测精确度分析
实验结果表明,苯酚、对苯二酚和对苯醌的富集倍数可达到75.0,72.7,69.1倍。做不同浓度富集实验,以富集后实际检测值去除富集倍数,得到相对误差,结果见表4。
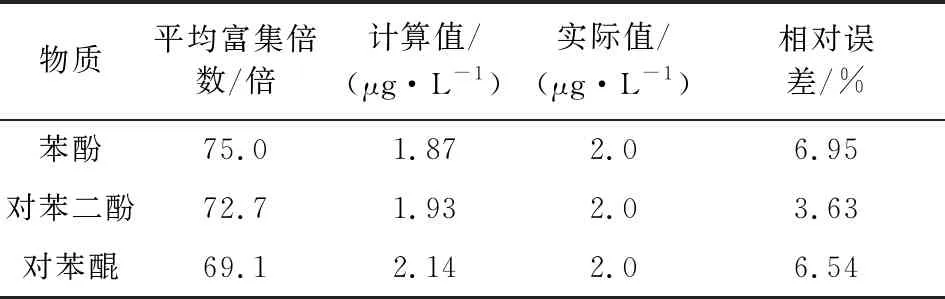
表4 检测分析误差
由表4可知,本实验相对误差在可接受范围内,3种物质分析精确度较高。
2.4 实例检测分析
分别在苯酚初始浓度为0.01,0.1,1 mg/L的情况下,以高铁酸钾与苯酚质量比为20∶1,进行高铁酸钾降解苯酚的实验,初始pH为9,反应时间为30 min, 反应温度为25 ℃。乙酸乙酯萃取,硫酸铵投加13 g,萃取时间20 min,液相色谱检测条件波长260 nm,流动相为甲醇∶水=40∶60,结果见表5。
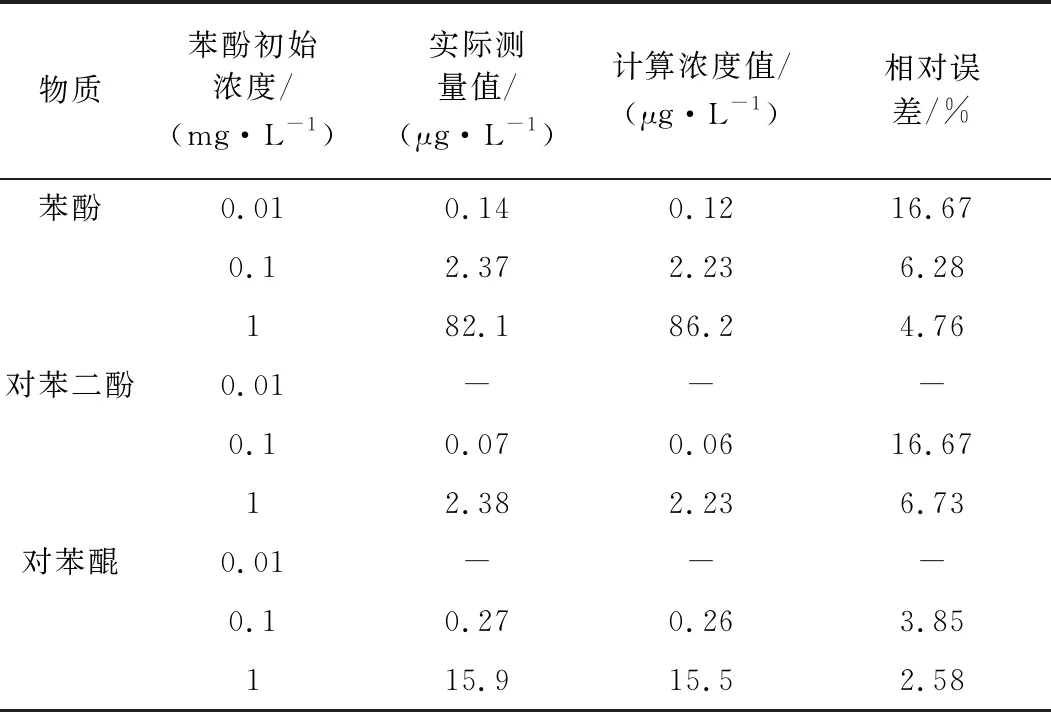
表5 实际检测效果
由表5可知,苯酚初始浓度在0.1 mg/L以上时,检测效果良好,但是初始浓度为0.01 mg/L时,由于反应产生的对苯二酚和对苯醌两种中间体过少,超出检测范围,无法准确检测,但是此时已足以说明苯酚降解后几种毒性强烈物质总量处于规范要求标准线下,满足给水厂水源水进厂要求。
3 结论
(1)未经过萃取的水样直接检测,杂峰多,不能满足高铁酸盐降解苯酚过程中微量残留苯酚、对苯二酚、对苯醌等有毒有害中间体检测的要求,通过氮吹浓缩的乙酸乙酯萃取是十分有效的液液萃取方法。
(2)HPLC色谱优化条件为波长260 nm,流动相为甲醇∶水=40∶60时,可有效消除物质峰不显示或重叠不能分离的情况,检测的残留苯酚、对苯二酚、对苯醌的物质峰分离明显,基本满足实验中对几种有毒有害物质的检测要求。
(3)盐析微萃取-液相色谱法联用法在合适的萃取和色谱条件下,可完成对苯酚等几种主要检测物质的富集检测,且实验精确度较高,是一种简易可靠的微量苯酚及其中间体浓度检测方法。
猜你喜欢
中国化肥信息(2022年6期)2023-01-06
化学分析计量(2021年6期)2021-06-22
云南化工(2020年11期)2021-01-14
中国化肥信息(2020年3期)2021-01-07
化学分析计量(2020年5期)2020-09-26
中国化肥信息(2018年8期)2018-10-08
中华皮肤科杂志(2018年6期)2018-01-19
山东化工(2017年5期)2017-09-16
合成化学(2015年4期)2016-01-17
化工生产与技术(2014年6期)2014-02-27
