
聚甘油单月桂酸酯-油酸微乳体系的构建及其性能
2021-05-19 07:05:24赵敬松王雨生李天昊赵文晴陈海华
食品科学 2021年8期
赵敬松,王雨生,2,李天昊,赵文晴,陈海华,*
(1.青岛农业大学食品科学与工程学院,山东 青岛 266109;2.青岛农业大学学报编辑部,山东 青岛 266109)
与乳状液相同,微乳液是由油相、水相、表面活性剂组成的,但本质上微乳液与乳状液完全不同。微乳液是自发形成的各向同性、外观透明或半透明的热力学稳定体系,具有更小的粒径(1~100 nm),稳定性更高。微乳液对水不溶性、油不溶性物质均有较强的增溶能力,因此可作为食品组分的载体,例如,可用于增加功能成分、色素等的溶解度。但食品体系的复杂性和安全性对食品级微乳液提出了更高的要求,而现有的微乳体系大都存在着安全性问题,或者不够高效,这很大程度上限制了微乳液在食品中的应用[1]。因此,亟需一种安全高效的微乳体系,以拓展微乳液在食品中的应用。
聚甘油单月桂酸酯是聚甘油与月桂酸形成的单酯,无色无味,对产品感官无不良影响,摄入人体后可被分解代谢,具有高度安全性,被联合国粮农组织(Food and Agriculture Organization,FAO)和世界卫生组织(World Health Organization,WHO)推荐使用[2],目前广泛应用在食品工业中[3]。然而作为一种安全高效的非离子型表面活性剂,聚甘油单月桂酸酯在微乳体系中的应用研究尚鲜见报道,其微乳制品的特性和稳定性也有待研究。
姜黄素是一种从植物中提取的含有二酮结构的色素,具有抗肿瘤[4]、调节免疫[5]、抗动脉粥样硬化[6]等多种生物活性,并具有一定的防腐作用[7],在食品工业中常用作着色剂。但姜黄素的水溶性差,生物利用率极低[8],生物活性容易受溶液的极性、表面活性剂的影响[9],这限制了姜黄素的应用。因此,提高姜黄素的溶解度和稳定性一直是近年来的研究热点。
本实验以构建安全高效的微乳体系、有效增溶姜黄素为目标,构建一种食品级可无限稀释的聚甘油单月桂酸酯-油酸微乳体系,探究表面活性剂与助表面活性剂质量比(Km)、水相pH值和NaCl浓度对微乳体系相行为的影响,分析微乳体系的结构,并对微乳液载姜黄素的性能进行研究,旨在提高姜黄素的溶解度,同时保持姜黄素的生物活性,拓展微乳液在食品中的应用范围。
1 材料与方法
1.1 材料与试剂
甘油单月桂酸酯(glycerol monolaurate,GML)、三聚甘油单月桂酸酯(triglycerol monolaurate,TGM)、六聚甘油单月桂酸酯(hexapolyglycerol laurate,HGM)、十聚甘油单月桂酸酯(decaglycerol laurate,DGM)山东滨州金盛新材料科技有限责任公司;姜黄素(分析纯) 北京索莱宝科技有限公司;聚乙二醇(聚合度400) 上海源叶生物科技有限公司;其他试剂均为国产分析纯。
1.2 仪器与设备
DF-101S集热式恒温加热磁力搅拌器 巩义市予华仪器有限责任公司;DDS-11C电导率仪 上海雷磁新泾仪器有限公司;ZEN3690激光粒度分析仪 英国马尔文仪器有限公司;UV-2000紫外-可见分光光度计 上海精密仪器仪表有限公司;HT7700透射电子显微镜 日立仪器有限公司。
1.3 方法
1.3.1 微乳液的制备
根据文献[10]的方法稍作修改。先将油相、表面活性剂和助表面活性剂按照一定比例混合均匀,然后在磁力搅拌器匀速搅拌下向体系中滴加去离子水,至体系澄清透明形成微乳液。
1.3.2 拟三元相图的绘制
按照表面活性剂与油相的质量比(surfactant to oily phase weight ratio,SOR)为10∶0、9∶1、8∶2、7∶3、6∶4、5∶5、4∶6、3∶7、2∶8、1∶9和0∶10配制混合体系,按照1.3.1节方法制备微乳液,记录体系由澄清变浑浊或由浑浊变澄清的临界点,并计算临界点时油相、表面活性剂和助表面活性剂相、水相三相各自所占比例,绘制拟三元相图,其中阴影区域为微乳相区,空白区域为非微乳相区,其分界线即为澄清和浑浊的临界点。
微乳液的可稀释能力,即最小可稀释比(dilution ratio,DR)可从拟三元相图直接获取[11-12]。图1中,过水相所在三角形顶点向对边引直线,直线上的点对应表面活性剂与油相之比,同一直线上SOR相等。该直线即为固定SOR的稀释线。拟三元相图就是沿不同SOR稀释线进行稀释的实验结果。当稀释线所经区域均为微乳相区时(图1中SOR=24∶1稀释线),说明在该SOR下无论如何改变水相占比,体系均为微乳,即可无限稀释。最小可稀释比是指当体系可无限度稀释时,所对应最小的SOR,因此,较小的DR表明形成可无限稀释体系所需表面活性剂比例较小。无DR即微乳体系不可无限稀释,记为DR=N/A。

图1 微乳液拟三元相图Fig. 1 Pseudo-ternary phase diagram of microemulsion
1.3.3 微乳液的最佳配方及影响因素
1.3.3.1 表面活性剂种类及复配比例
固定制备温度为25 ℃,选择不同的表面活性剂,以乙醇作为助表面活性剂,表面活性剂与助表面活性剂质量比Km=1,制备微乳液并绘制拟三元相图。
1.3.3.2 助表面活性剂种类
固定制备温度为25 ℃,表面活性剂为HGM和DGM的复配物(质量比为2∶1),助表面活性剂分别为乙醇、丙二醇、丙三醇、聚乙二醇,Km=1,制备微乳液并绘制拟三元相图。
1.3.3.3Km值
固定制备温度为25 ℃,表面活性剂为HGM和DGM的复配物(质量比为2∶1),以乙醇作为助表面活性剂,Km分别为2/1、1/1、1/2、1/3,制备微乳液并绘制拟三元相图。
1.3.3.4 水相pH值
固定制备温度为25 ℃,表面活性剂为HGM和DGM的复配物(质量比为2∶1),以乙醇作为助表面活性剂,Km=1,水相pH值分别为3、5、7、9、11,制备微乳液并绘制拟三元相图。
1.3.3.5 水相NaCl浓度
固定制备温度为25 ℃,表面活性剂为HGM和DGM的复配物(质量比为2∶1),以乙醇作为助表面活性剂,Km=1,水相NaCl浓度分别为0.2、0.4、0.6、0.8、1.0、1.2 mol/L,制备微乳液并绘制拟三元相图。
1.3.4 微乳液粒径的测定
将制备好的微乳液样品加入清洁的样品池中,25 ℃恒温保持2 min,使用激光粒度分析仪测定微乳体系粒径。
1.3.5 微乳体系结构表征
采用电导率法[13]测定微乳结构。将表面活性剂、助表面活性剂和油相充分混合,并向其中逐滴滴加去离子水,记录体系电导率。
1.3.6 微乳液的微观形貌观察
将制备好的微乳液样品滴加在铜网上,冻干后置于透射电镜观察。
1.3.7 载姜黄素微乳液性能表征
1.3.7.1 姜黄素在微乳液中的溶解度
准确称取姜黄素标准样品6 mg,溶于100 mL 95%乙醇溶液中。分别取250、500、1 000、1 500、2 000 μL上述溶液用95%乙醇溶液定容至15 mL,配制成质量浓度分别为0.001、0.002、0.004、0.006、0.008 mg/mL的标准溶液,于425 nm波长处测定吸光度(Y),得到姜黄素质量浓度(X/(mg/mL))标准曲线Y=134.54X+0.003 1,R2=0.999 8。取适量姜黄素微乳液稀释于300 倍体积的95%乙醇溶液中,测定其吸光度,并根据质量浓度标准曲线计算姜黄素溶解度。
1.3.7.2 1,1-二苯基-2-三硝基苯肼(1,1-diphenyl-2-trinitrophenylhydrazine,DPPH)自由基清除能力
参照Wang Bini等[14]方法并稍做修改,测定姜黄素乙醇溶液、姜黄素微乳液对DPPH自由基的清除能力。用无水乙醇溶解DPPH并制备成0.1 mmol/L的储备液。取1 mL样品液,加入至2 mL DPPH储备液,涡旋均匀后,于室温下避光反应30 min,以无水乙醇为空白,在517 nm波长处测定吸光度为Ai。将1 mL样品液加入至2 mL无水乙醇中,测定吸光度为Aj。将1 mL无水乙醇加入至2 mL DPPH储备液中,测定吸光度为Ac,则待测液的DPPH自由基清除率如下:

1.4 数据处理
每个样品平行测定3 次,取平均值。利用SPSS V22.0进行显著性分析。采用Origin 8.0软件绘制拟三元相图,采用AutoCAD 2014计算DR及微乳区域面积占比S。
2 结果与分析
2.1 微乳体系的构建
2.1.1 表面活性剂的筛选
聚甘油单月桂酸酯分子中含有大量的羟基,聚合度不同的分子,其亲水链的长度、羟基数量不同,直接影响亲水亲油平衡值(hydrophilic-lipophilic balance,HLB)和微乳形成能力[15]。
由图2可知,随着聚甘油单月桂酸酯聚合度的增大,微乳区域面积占比S先增大后减小,以HGM作为表面活性剂时,制备的微乳体系S最大,达到30.05%,而以GML为表面活性剂时,S最小。这表明聚甘油单月桂酸酯的聚合度对微乳液的形成有影响。这是因为当聚合度较低时,聚甘油单月桂酸酯分子链上的羟基较少,与水相之间可形成氢键的数量较少,载水量较低,另外分子链较短,亲油性较弱[16],形成微乳的能力较差,表现为S较小。聚合度的升高又会引起HLB的升高,而过高的HLB将导致表面活性剂分子难以在水相与油相之间相互穿插形成胶束,从而降低微乳液的形成能力,表现为S减小[17]。
由图2还可以看出,采用GML和TGM制备的微乳液没有DR,即采用这两种表面活性剂时,表面活性剂和油相在任何配比下都无法形成可无限稀释体系[18]。与DGM相比,采用HGM制备的微乳液,DR较小,说明乳化相同含量的油相并形成可无限稀释体系,所需表面活性剂较少。本实验选择能够形成可无限稀释微乳体系的HGM、DGM进行复配形成表面活性剂。
配伍性较好的表面活性剂存在协同增效作用,复配后可以提高乳化效率,降低表面活性剂分子空间位阻,增加油水界面的柔性,提高微乳液的稳定性[19]。如图3所示,HGM与DGM以不同比例复配,随着体系中HGM含量的增多,S先增大后减小。当HGM和DGM质量比为2∶1时,S最大为30.08%,与单一HGM作为表面活性剂时的S一致。另外,DR=9,显著低于单一HGM和单一DGM的DR。这表明HGM与DGM间存在协同增效作用,复配后可以有效提高乳化能力,容易形成可无限稀释微乳体系。因此,本实验将HGM和DGM以质量比为2∶1的复配物作为表面活性剂。
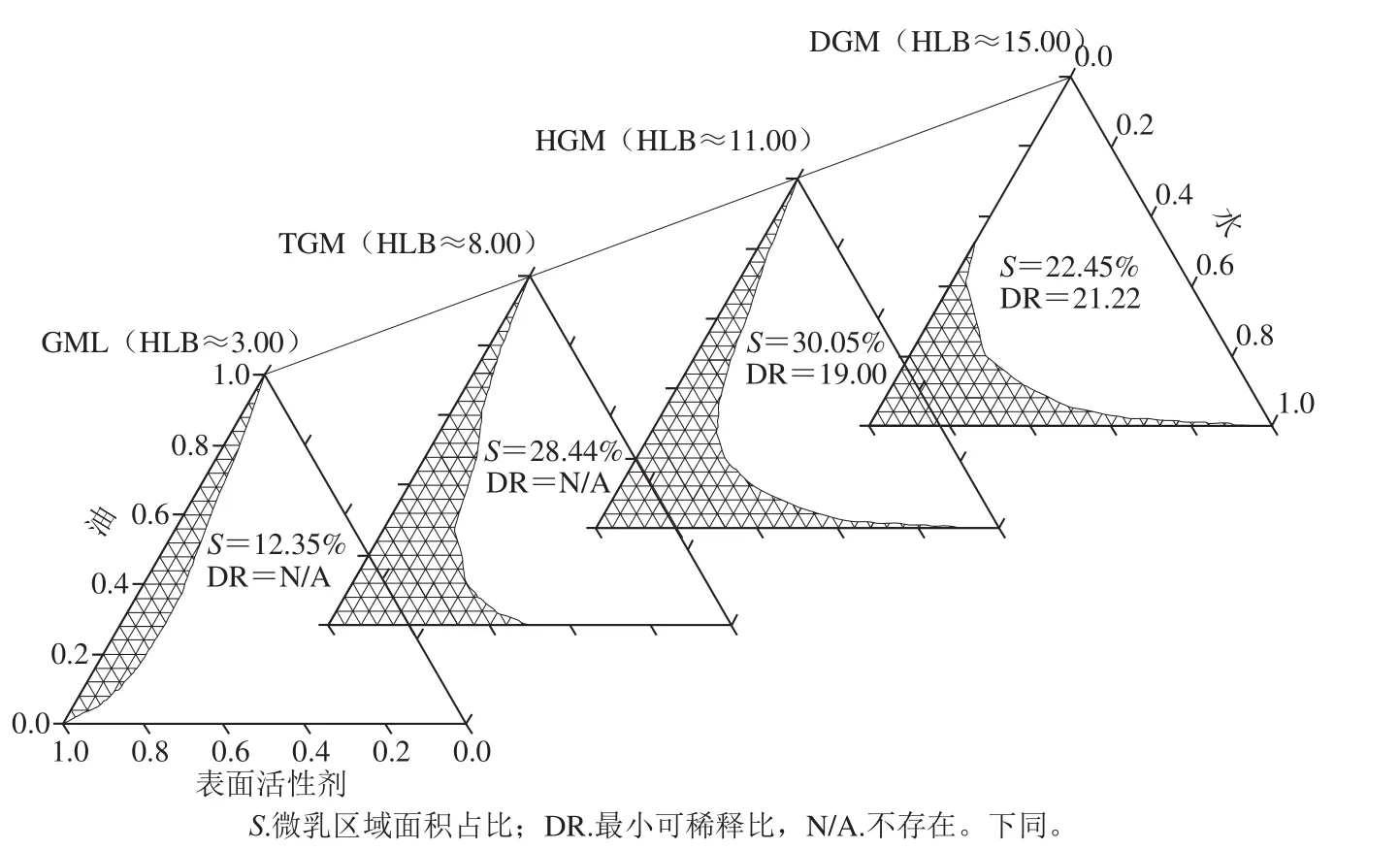
图2 不同聚合度的聚甘油单月桂酸酯制备微乳液的拟三元相图Fig. 2 Pseudo-ternary phase diagrams of microemulsions prepared with polyglycerol monosaurates with different degrees of polymerization
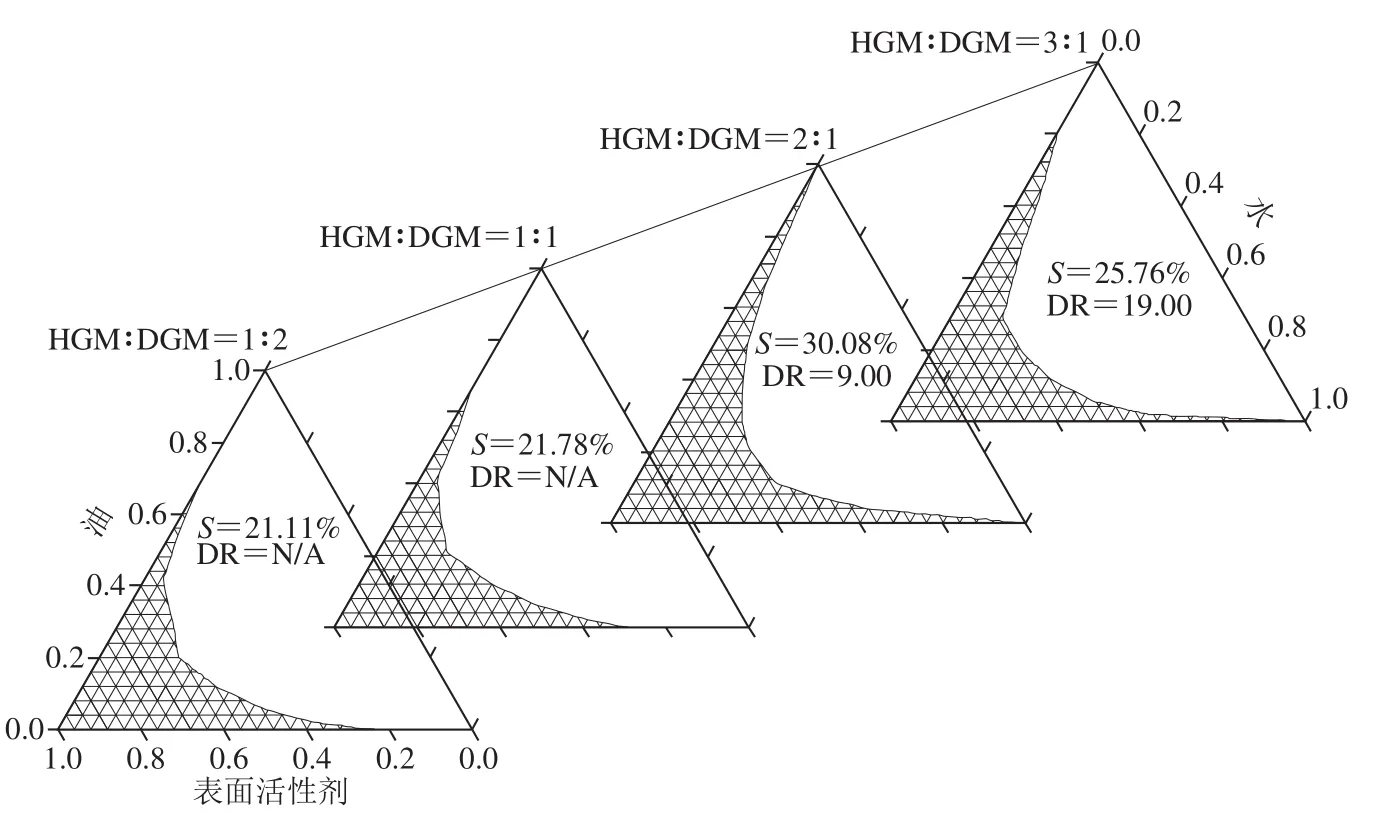
图3 HGM与DGM不同复配比例制备微乳液的拟三元相图Fig. 3 Pseudo-ternary phase diagrams of microemulsions prepared with different mixing ratios between HGM and DGM
2.1.2 助表面活性剂的筛选
在微乳液的制备中,除表面活性剂外,通常还需要添加助表面活性剂进一步降低体系的表面张力。助表面活性剂可嵌入至表面活性剂分子间,从而降低表面活性剂分子间的相互作用力和界面弯曲能,增强界面膜的流动性,利于微乳液的形成[20]。此外,助表面活性剂还可以充当助溶剂,对外来物质起增溶作用[21]。
由图4可知,4 种助表面活性剂中,乙醇作为助表面活性剂时形成的微乳体系澄清透明,流动性好,S最大,DR=9,是唯一能形成可无限稀释微乳体系的助表面活性剂。而丙二醇、丙三醇、聚乙二醇所形成的体系黏稠、流动性差,临界点不易判断,甚至会有分层现象,S明显较小。朱加进等[22]研究发现,使用乙醇、丙二醇、丙三醇作为助表面活性剂制备磷虾油微乳液时,丙三醇无法形成微乳液,乙醇和丙二醇可以形成,且使用乙醇时S最大。Chaiyana等[23]分别采用乙醇、己醇制备微乳液时发现,乙醇体系的S远大于己醇。邹华生等[24]通过分析拟三元相图得出,分别采用乙醇、正丁醇、正戊醇、异丁醇制备环己烷微乳液时,所得到S依次减小,且采用正戊醇和异丁醇时体系黏度明显上升。乙醇作为助表面活性剂制备微乳液时S较大,这可能是由于乙醇分子空间位阻较小,更容易嵌入表面活性剂分子间,使吸附在界面膜上的乳化粒子排列更加紧密[25],从而降低了体系黏度,利于微乳液的形成[26];而丙二醇、丙三醇、聚乙二醇的分子链较长,亲油能力强,甚至可充当油相角色,难以分配到油水界面上,导致形成微乳液的能力下降[27]。因此,选取乙醇作为助表面活性剂。
2.1.3Km的筛选
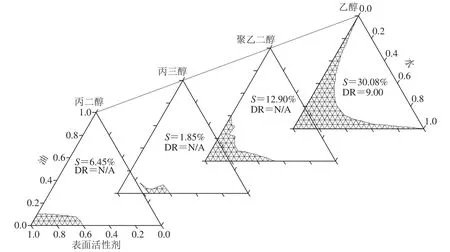
图4 不同助表面活性剂制备微乳液的拟三元相图Fig. 4 Pseudo-ternary phase diagrams of microemulsions prepared with different cosurfactants

图5 不同Km条件制备微乳液的拟三元相图Fig. 5 Pseudo-ternary phase diagrams of microemulsions prepared with different Km values
如图5所示,Km对微乳区域面积影响显著,随着Km的减小,S逐渐升高。这可能是由于降低Km会使更多的助表面活性剂渗透到表面活性剂界面膜中,这增加了界面膜的柔性,有利于微乳液的形成[28],另外,助表面活性剂亲水性强,增水量较大,会进一步增大S。随着Km的减小,DR、d均先降低后升高,且Km=1/2时最低。这可能是由于Km较大时,助表面活性剂含量较低,增水量较小,界面膜难以弯曲,表现为较大的DR和d。而当Km过小时,过量的助表面活性剂使表面活性剂从油水界面向连续相迁移[29],难以维持较低的油水界面张力,水分含量较高时界面容易失去平衡,导致DR和d明显升高[30]。因此,选取Km=1/2制备微乳液。
2.1.4 水相pH值的影响
由图6可知,随着水相pH值升高,S先升高后降低,在pH 7时,S最大;DR和d则先降低后升高,pH 7时最低。这表明水相pH值小于或大于7都不利于微乳液的形成,形成的微乳液粒径较大。陶紫等[31]研究发现,体系的pH值越偏离中性,其电导率越大,油-水界面膜越不稳定,微乳液越容易失稳。

图6 不同pH值水相制备微乳液的拟三元相图Fig. 6 Pseudo-ternary phase diagrams of microemulsions prepared with aqueous phases at different pH values
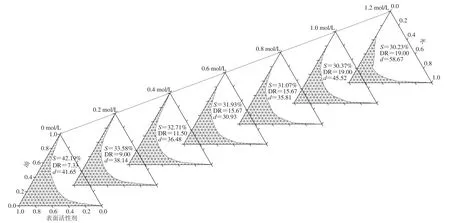
图7 不同NaCl浓度水相制备微乳液的拟三元相图Fig. 7 Pseudo-ternary phase diagrams of microemulsions prepared with aqueous phases with different NaCl concentrations
2.1.5 NaCl浓度的影响
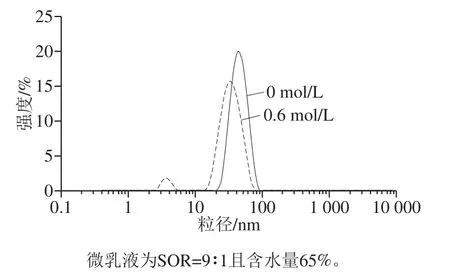
图8 不同NaCl浓度水相制备微乳液的粒径分布Fig. 8 Particle size distribution of microemulsions prepared with aqueous phase with different NaCl concentrations
如图7所示,随着水相中NaCl浓度的逐渐升高,S明显减小,DR升高。这可能是由于电解质浓度升高逐渐增强了表面活性剂的亲油性[32],使体系的增水量逐渐降低,形成可无限稀释微乳体系的难度逐渐加大。而较高的离子强度会使表面活性剂发生盐析,导致体系中游离的表面活性剂减少,难以维持较低的表面张力,从而抑制了微乳的形成[1]。张佩华等[33]根据实验推测得出类似结果:随着加入的电解质的增多,表面活性剂亲油性增强、亲水性减弱,体系增溶能力下降。值得注意的是,随着水相中NaCl浓度的升高,微乳粒径先降低后升高,NaCl浓度为0.6 mol/L时,微乳粒径最小,为30.93 nm。这可能是由于少量的电解质加入使胶束双电层压缩,促使体系液滴进一步细化[1],粒径减小。另外,电解质可以降低表面活性剂的临界胶束浓度,使部分表面活性剂自身发生缔合形成胶束,如图8中1~10 nm处的峰即为胶束峰(10~100 nm处的峰为微乳液滴粒径分布峰)。胶束的粒径较小,胶束的出现降低了体系平均粒径。当水相中NaCl浓度过高时,液滴双电层被进一步压缩,液滴间容易互相接近,产生聚结,导致粒径增大。
2.2 微乳液的结构表征
2.2.1 SOR对微乳液粒径的影响
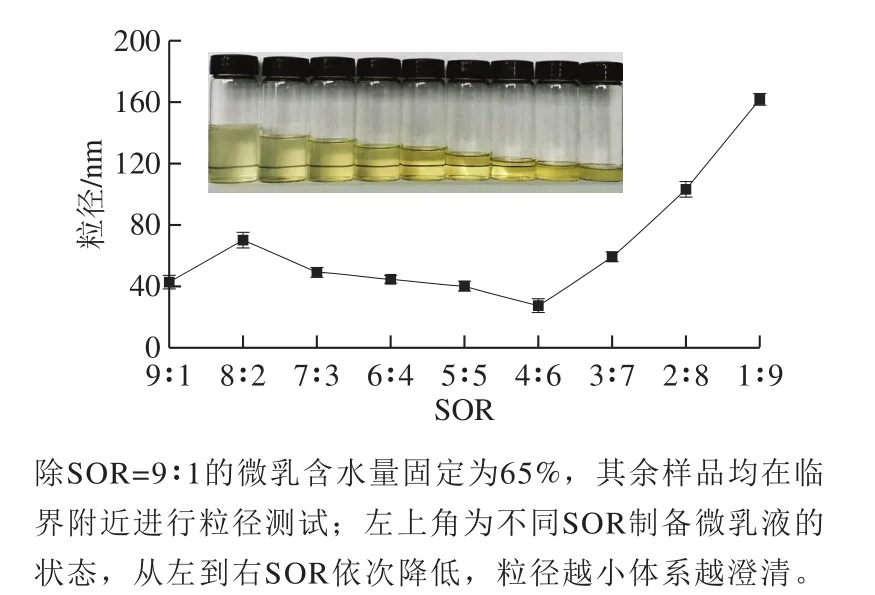
图9 不同SOR制备的微乳液粒径Fig. 9 Particle size of microemulsions prepared with different SORs at critical point
从图9可以看出,SOR大于2∶8时,粒径均小于100 nm,体系澄清透明。随着SOR的降低,微乳液的粒径先略有减小后增大,SOR为4∶6时,粒径最小,为26.69 nm。当SOR小于3∶7时,较少的表面活性剂难以维持较低的表面张力,导致微乳液粒径增大,体系变得浑浊[34]。此外,实验中发现,SOR=8∶2时,制备的微乳液黏度较高,这或许是该条件下制备的微乳液粒径较大原因。
2.2.2 微乳液结构分析
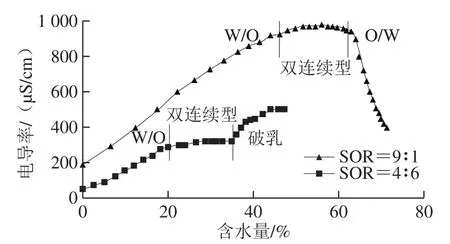
图10 微乳液电导率随水分含量变化的曲线Fig. 10 Change in electrical conductivity of microemulsions with water content
由图10可知,SOR=9∶1时,微乳液可无限稀释,即为无限可稀释微乳体系。随着含水量的升高,微乳液的电导率变化分为3 个阶段,第1阶段为含水量较小(低于45%)的阶段,在该阶段,随着含水量的增加,电导率迅速上升,这说明微乳液为油包水型(W/O型),通过分散于油相中的水滴互相碰撞而导电。W/O型微乳液的含水量升高时,其中的水滴密度随之升高,水滴来回碰撞机会增多,形成导电链,表现为电导率升高[35]。第2阶段微乳液的含水量介于45%和60%间,在该阶段,提高微乳液的含水量,电导率变化较慢,这说明微乳液已由W/O型转变为双连续型,液滴间发生黏性碰撞,油相和水相相互交错为网络通道,均形成连续相,因此提高含水量对微乳液的电导率影响不大[36]。第3阶段的含水量超过60%,在该阶段,提高微乳液的含水量,电导率迅速下降,说明微乳液由双连续型完全转变为水包油型(O/W型),O/W型微乳液的含水量增加时,作为导电粒子的油滴被逐渐稀释,电导率下降[37]。
李玉琴等[38]以油酸为油相,以吐温80为表面活性剂,以无水乙醇为助助表面活性剂,在SOR为9∶1时,仅能形成W/O型和双连续型微乳液,且不可无限稀释。郭静等[30]将大豆卵磷脂、Span80与无水乙醇按照质量比3∶1∶2混合为表面活性剂相,以甘油溶液为水相,在SOR为9∶1时,增水量超过30%时就会破乳。因此本实验所制备的聚甘油单月桂酸酯-油酸微乳体系具有良好的增水能力及可稀释性。
图10中,SOR为4∶6的微乳液,其第1阶段和第2阶段(含水量分别为低于20%、介于20%和35%之间)的电导率的变化趋势与SOR=9∶1的微乳液一致,但第3阶段(含水量超过35%)电导率随含水量的提高而迅速上升,这可能是过多的水分破坏了微乳液的界面膜,发生了破乳现象。这说明SOR=4∶6时,微乳液不能转变为O/W型。
2.2.3 微乳液的微观形貌

图11 微乳液的透射电镜照片Fig. 11 Transmission electron microscope photos of microemulsion
从图11可以看出,微乳液滴呈均匀球形,表面光滑,平均粒径10~100 nm。这与2.1.5节所测的微乳液粒径分布一致,表明体系已形成均一的O/W型微乳液。
2.3 载姜黄素微乳液的特性评价
2.3.1 姜黄素溶解度

表1 姜黄素在不同溶剂中的溶解度Table 1 Solubility of curcumin in different solvents
由表1可知,姜黄素在油酸中的溶解度最低,仅为0.299 mg/mL,在质量分数均为15%的HGM和DGM表面活性剂水溶液中,姜黄素的溶解度有所升高,但仍很小,而在相同表面活性剂浓度的微乳液中,姜黄素溶解度明显提高,可达到4.874 mg/mL,这说明微乳液具有比单一组分更高的溶解能力。曾庆晗等[39]以中链甘油三酯和卵磷脂制备了纳米乳,姜黄素溶解度为3.669 mg/mL;匡建[40]以Gemini双子表面活性剂制备的微乳液,在SOR=9∶1时,姜黄素的溶解度为4.709 mg/g,但Gemini双子表面活性剂不允许应用在食品中。此外,烷基酚聚氧乙烯醚和聚氧乙烯氢化蓖麻油被广泛报道用于制备微乳液增溶营养素[41-42]但均存在一定的安全性问题,在食品中的应用受到限制。另外,采用普通有机溶剂溶解姜黄素,在稀释后易析出或沉淀,而采用可无限稀释的水包油型微乳体系,由于姜黄素溶解在油相中,稀释只会扩大连续相(水相)体积,并不改变油滴结构[12],稳定性更好。因此,本实验所得到的聚甘油单月桂酸酯-油酸微乳体系安全性高、稳定性好,能够有效提高姜黄素溶解度。
2.3.2 DPPH自由基清除能力
采用SOR=9∶1制备微乳液,调节含水量至65%,以此作为载体,测定不同姜黄素添加量微乳液的DPPH自由基清除能力。以姜黄素乙醇溶液作为对照。

图12 姜黄素微乳液及姜黄素乙醇溶液的DPPH自由基清除能力Fig. 12 Scavenging effects of curcumin microemulsion and curcumin ethanol solution on DPPH radical
由图12可知,姜黄素乙醇溶液的DPPH自由基清除率随着姜黄素添加量的增加而上升。当姜黄素添加量超过0.15 mg/g时,继续提高姜黄素添加量,DPPH自由基清除率不再变化。而姜黄素微乳液的DPPH自由基清除率变化趋势与姜黄素乙醇溶液相似。姜黄素质量浓度相同时,姜黄素微乳液的DPPH自由基清除率接近于姜黄素乙醇溶液的,差异很小。这表明姜黄素微乳化并不影响姜黄素的生物活性,本实验所制备微乳液可作为姜黄素的优良载体。
3 结 论
本实验使用聚甘油月桂酸酯和油酸,通过拟三元相图法确定了一种安全性高、可稀释的微乳体系配方,并探究水相pH值、NaCl浓度对微乳体系的影响,采用电导率法测定了体系的相转变行为,最后分析了微乳体系载姜黄素性能。微乳液的配方为:HGM和DGM按质量比2∶1复配为表面活性剂,乙醇为助表面活性剂,Km=1/2。该微乳体系具有42.19%的最大微乳区面积,最小可稀释比低至7.33,表面活性剂比例较低。当体系含水量超过60%时,可形成O/W型均一微乳液。激光粒度分析及透射电镜照片显示,微乳液滴呈均一球形,粒径最小为26.69 nm,证实了微乳的形成。该微乳体系具有较高的载姜黄素能力,姜黄素溶解度达4.87 mg/mL,溶解在该微乳体系的姜黄素抗氧化活性与乙醇姜黄素溶液相当,保持了姜黄素的生物活性,有效解决了姜黄素水溶性差、稀释性差问题。关于微乳体系对其他疏水物质的运载性能尚需进一步研究。
猜你喜欢
能源化工(2022年3期)2023-01-15 02:26:43
现代畜牧科技(2021年8期)2021-10-13 07:21:42
石油地质与工程(2019年3期)2019-09-10 08:27:54
中国钼业(2019年2期)2019-01-19 15:54:06
中成药(2017年7期)2017-11-22 07:32:54
水利技术监督(2016年6期)2017-01-15 14:01:33
无机化学学报(2014年6期)2014-07-14 05:19:42
天然产物研究与开发(2014年8期)2014-04-27 14:16:36
湿法冶金(2014年3期)2014-04-08 01:04:51
食品与生物技术学报(2014年11期)2014-04-07 00:28:29
