
N6-甲基腺嘌呤去甲基化酶FTO和ALKBH5在肿瘤中的研究进展
2021-05-17 13:18何宜宸陈依梦吴昌平
肿瘤 2021年4期
何宜宸,陈依梦,吴昌平
基因的表达调控体现在转录调控、转录后修饰和翻译调控等多个层面,这共同构成了机体复杂而精密的调控网络。目前,已知的转录后修饰多达163种[1]。这些修饰不仅影响了RNA的分子结构,改变了其稳定性,同时也使其获得了更多的生物学功能,增加了RNA的多样性。在大多数真核生物mRNA中,RNA腺苷N6位上的甲基化修饰(N6-methyladenosine,m6A)是最为常见的转录后修饰形式[2]。尽管m6A修饰极为普遍,然而当前对其在各种疾病中发挥的生物学功能却知之甚少[3]。m6A修饰最常见的识别基序(motif)为RRm6ACH [(G/A/U)(G/A)m6AC(U/A/C)],且通常富集于终止密码子以及RNA 3’端和5’端非翻译区附近[4-5]。m6A修饰对前体RNA剪接、RNA 5’和3’端加工、RNA出核、RNA代谢以及RNA稳定性和翻译水平调控都发挥着至关重要的作用[6-14]。
m6A修饰的主要相关蛋白包括“书写器”(writter)、“擦除器”(eraser)和“阅读器”(reader),这些蛋白分别能够添加、移除和识别m6A修饰位点。其中,m6A“书写器”为一类多组分的甲基转移酶复合物,包括甲基化转移酶3蛋白(methyltransferase-like protein 3,METTL3)和甲基化转移酶14蛋白(methyltransferase-like protein 14,METTL14)以及其辅助因子人肾母细胞瘤1关联蛋白(Wilms’ tumor 1-associated protein,WTAP)、RNA结合基序蛋白15/15B(RNA binding motif protein 15/15B,RBM15/15B)、类病毒m6A甲基转移酶相关蛋白VIRMA(KIAA1429)和锌指结构域包含蛋白13(Zinc finger CCCH domain-containing protein 13,ZC3H13)等[15-19]。2011年,在JIA等[20]首次发现脂肪和肥胖相关基因蛋白(fat mass and obesity-associated protein,FTO)能够扮演m6A“擦除器”的角色后,m6A修饰被认为是可逆的且常处于动态调节状态中。随后,ZHENG等[21]在2013年研究小鼠的生育功能时发现了新的m6A“擦除器”——AlkB同源蛋白5(AlkB homologue 5,ALKBH5)。目前已知的阅读蛋白包括YT521-B同 源(YT521-B homology,YTH)结构域蛋白家族(YTHDF1、YTHDF2、YTHDF3、YTHDC1和YTHDC2)、胰岛素样生长因子2 mRNA结合蛋白(insulin-like growth factor 2 mRNA binding protein,IGF2BP)家族(IGF2BP1、IGF2BP2和IGF2BP3)以 及核不均一核糖核蛋白(heterogeneous nuclear ribonucleoprotein,HNRNP)家族(HNRNPA2B1、HNRNPC和HNRNPG)[3,9,22-26]。在m6A“书写器”和“擦除器”动态调节m6A修饰水平后,各种“阅读器”蛋白随后即进行修饰位点的识别,发挥多种调控作用,影响多种生物学功能,包括但不限于胚胎发育、性别决定和应激反应等[27-28]。由于m6A修饰参与调节了多种正常生理活动,m6A关键酶的异常或m6A修饰水平的异常常会引发多种疾病,如神经性疾病、免疫缺陷和多种肿瘤[29-31]。本文旨在探讨肿瘤病理状态对m6A“擦除器”FTO和ALKBH5与肿瘤发生和进展的关系,并将就其分子机制作一综述(表1)。
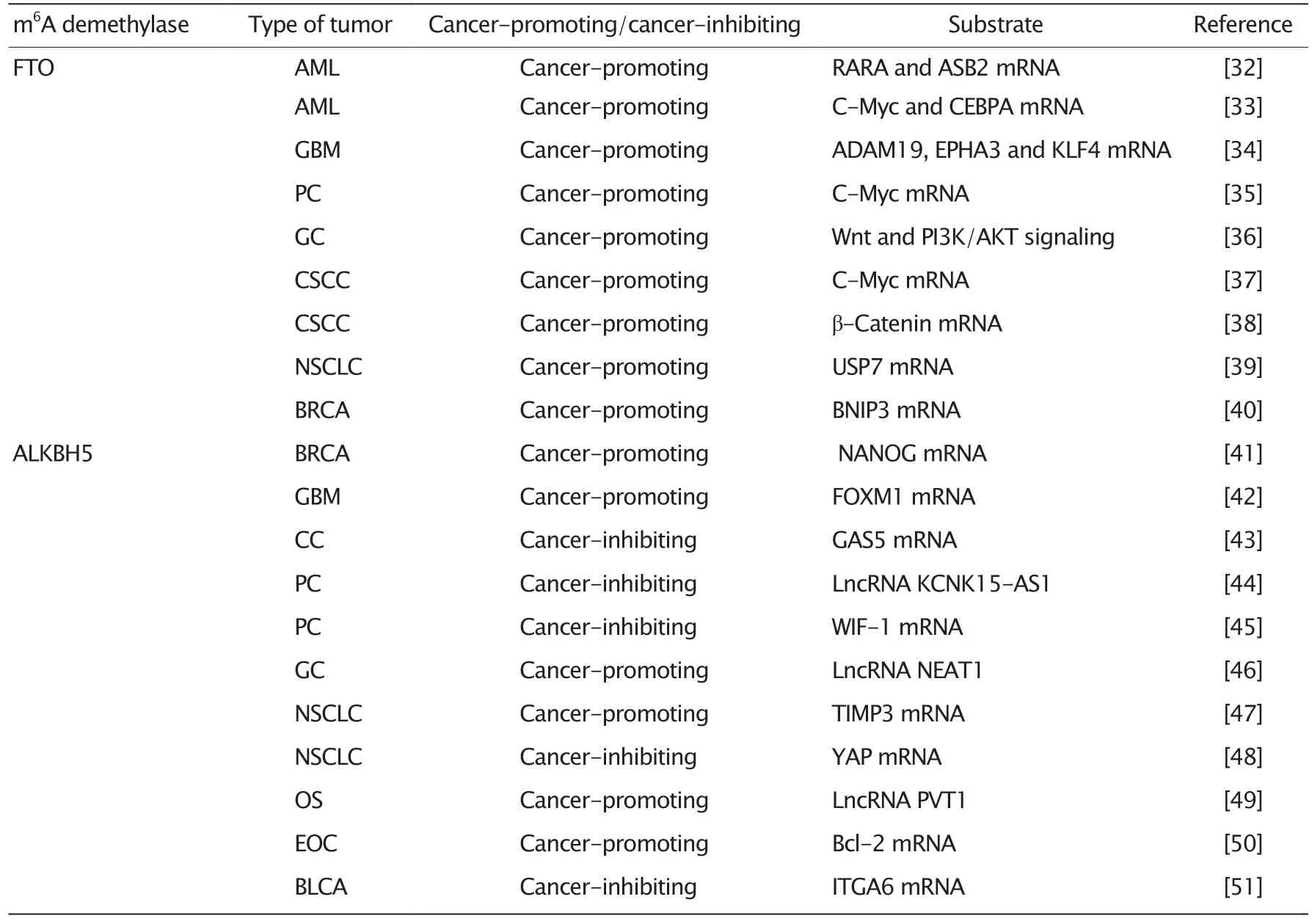
表1 N6甲基腺嘌呤去甲基化酶在多种肿瘤中的作用Table 1 Role of m6A demethylases in multiple tumor types
FTO基因最早由VAN DER HOEVEN等[52]在发生突变的融合脚趾小鼠8号染色体上发现,并被认为与细胞的程序性凋亡有关。在人类基因组中,FTO基因位于人类16号染色体长臂12区2带,具有9个外显子,在人类不同发育阶段的多种组织内表达,在大脑中有极高的表达量[53-54]。FTO基因被发现能够编码2-酮戊二酸(2-oxoglutarate,2-OG)依赖的核酸去甲基化酶,催化单链DNA上的3-甲基胸腺嘧啶脱甲基[55],但随后单链RNA上的m6A修饰被证明是其主要底物[20]。FTO被认为通过动态调节m6A修饰水平参与了糖脂代谢、神经传导缺陷修复以及乳腺癌、肝癌和肺癌等多种恶性肿瘤的发生和进展[11,39-40,56-59]。
2013年,ZHENG等[21]在FTO的同源家族中发现了第2种m6A去甲基化酶ALKBH5。ALKBH5和FTO均属于Fe2+和2-OG依赖的AlkB双加氧酶家族。该家族共有9个成员,分别为FTO和ALKBH1~8。ALKBH5被发现可以调控单链RNA和单链DNA上的甲基化修饰[60]。THALHAMMER等[61]发现在多种细胞系中,缺氧环境可以激活ALKBH5基因的转录,ALKBH5是缺氧诱导因子1(hypoxia inducible factor-1,HIF-1)的下游靶基因,可参与细胞对于缺氧微环境的反应。考虑到恶性肿瘤中极为常见的肿瘤缺氧微环境,ALKBH5可能通过此方式参与了恶性肿瘤的进展。尽管具体机制尚未被完全阐明,ALKBH5与乳腺癌、胃癌和白血病等多种恶性肿瘤的密切关系已被报道[46,62-63]。
1 FTO与肿瘤
1.1 FTO与急性髓系白血病
FTO在急性髓系白血病的部分亚型中高表达,并在其中扮演促癌蛋白的角色。维甲酸受体α(retinoic acid receptor α,RARA)以及人锚蛋白重复和SOCS框蛋白2(ankyrin repeat and SOCS box containing 2,ASB2)被证明是急性髓系白血病中的抑癌基因,而FTO能够去除其mRNA上的m6A修饰并诱导其降解,从而抑制其表达[64],高表达的FTO被报道能促进急性髓系白血病的进展[32,65]。此外,由于FTO/RARA/ASB2轴在RARA诱导的促白血病细胞分化效应中的关键作用,联合FTO抑制剂与全反式维甲酸(all-trans retinoic acid,ATRA)诱导剂的治疗方案已被证明对FTO高表达的急性髓系白血病亚型有着良好的疗效[64]。
FTO/C-Myc/CCAAT增强子结合蛋白α(CCAAT enhancer binding protein α,CEBPA)通路是FTO在白血病进展中的另一条信号通路,FTO在此通路中同样被认为是通过调控m6A修饰而影响下游基因。FTO在去除原癌基因C-Myc mRNA上的修饰后,可以增加其稳定性而上调其表达水平,最终导致白血病进展加速。另外,在急性髓系白血病中,FTO可以通过调节CEBPA mRNA的m6A修饰水平而增加其表达,而异常增高的CEBPA可作用于FTO基因的增强子,促进其转录,从而形成正反馈回路[33]。FTO抑制剂R-2-羟基戊二酸(R-2-hydroxyglutarate,R-2HG)被发现可以通过阻断FTO/C-Myc/CEBPA通路而发挥抑癌作用,其在FTO高表达的急性髓系白血病亚型中具有广阔的临床应用前景[33]。
1.2 FTO与胶质母细胞瘤
肿瘤干细胞被认为可能是恶性肿瘤中所有肿瘤细胞的起源[66],其不仅具有自我更新能力,还可分化出异质性的子代肿瘤细胞,对肿瘤细胞的增值、侵袭能力以及药物抵抗性产生巨大影响。m6A修饰水平的改变被证明可以影响胶质母细胞瘤干细胞(glioblastoma stem cells,GSCs)的生长和自我更新,较低的m6A修饰水平与胶质母细胞瘤的进展和不良预后相关[34]。m6A去甲基化酶FTO可通过降低m6A修饰水平而导致癌基因ADAM金属肽酶域19(ADAM metallopeptidase domain 19,ADAM19)、EPH受 体A3(EPH receptor A3,EPHA3)以 及Kruppel样因子4(Kruppel-like factor 4,KLF4)的表达上调,并促进GSCs生长和自我更新,从而影响肿瘤的发生、进展以及放化疗抵抗[34]。
新发现的FTO抑制剂甲氯芬那酸的乙酯衍生物2(meclofenamic acid 2,MA2)被证明可抑制由GSCs引起的胶质母细胞瘤的发生,并可使已接种GSCs的小鼠生存期延长[34]。
1.3 FTO与胰腺癌
在胰腺癌中,原癌基因C-Myc的转录物被认为是FTO进行m6A修饰水平调节的主要底物。FTO被发现可以去除C-Myc mRNA上的m6A修饰,进而增加C-Myc的稳定表达,并促进胰腺癌细胞的增殖[35]。然而,FTO作为“脂肪和肥胖相关基因”,与脂肪代谢调节以及身体质量指数也有着密切联系[67],而过高的体质量和高脂饮食正是是胰腺癌的高危因素[68-69]。因此,通过调节C-Myc mRNA的m6A修饰水平而在胰腺癌中发挥促癌效应,可能只是FTO在影响胰腺癌进展的多种机制之一。
1.4 FTO与胃癌
FTO基因在胃癌组织中呈高表达,并且其表达量与胃癌病理分级和患者预后密切相关[70]。FTO基因在发生远处转移、高TNM分期以及低分化患者的胃癌组织中均被观察到高表达,并被认为可作为独立的预后影响因子[70]。同时,上调FTO表达水平可观察到胃癌细胞增殖、迁移和侵袭的加速,而下调FTO表达水平则相反[71]。
在分子机制方面,m6A修饰水平被认为在胃癌进展中有着决定性的作用,m6A“书写器”和“阅读器”是潜在的抑癌蛋白,而m6A“擦除器”则发挥着促癌效应[36]。m6A“擦除器”FTO可通过降低m6A修饰水平来激活无翅型MMTV整合位点家族(wingless-type MMTV integration site family,Wnt)和磷脂酰肌醇3激酶(phosphoinositide 3 kinase,PI3K)/蛋白激酶B(protein kinase B,PKB,又称AKT)信号通路,从而促进胃癌细胞的增殖和侵袭。
在临床研究中,胃癌患者外周血样本中全血的总RNA m6A修饰水平被发现增高,而FTO表达降低。另外,通过统计胃癌患者与健康对照者m6A修饰水平数据绘制受试者工作特征曲线(receiver operating characteristic curve,ROC),m6A修饰水平被认为可以作为胃癌患者的非侵入性诊断指标,其诊断效率达到了92.9%,远高于胃肠道肿瘤标志物肿瘤相关糖类抗原(carbohydrate antigen 19-9,CA19-9)(60.3%)和癌胚抗原(carcinoembryonic antigen,CEA)(69.4%)的诊断效率[72]。
1.5 FTO与宫颈鳞状细胞癌
同急性髓系白血病类似,FTO在宫颈鳞状细胞癌中也被认为通过上调C-Myc基因而促进了肿瘤细胞的增殖和侵袭。通过引入去甲基化酶活性位点突变的FTO功能缺失型突变体作为对照组,FTO上调C-Myc基因表达的机制同样被认为是通过去除其mRNA的m6A修饰而增加C-Myc mRNA的稳定性。在FTO基因敲除的宫颈癌细胞系中,将C-Myc基因过表达可逆转FTO基因敲除造成的细胞增殖减慢和侵袭能力下降效应[37]。
另外,m6A去甲基化酶FTO还可通过影响m6A修饰水平来调节宫颈鳞状细胞癌的放化疗抵抗性,在宫颈鳞状细胞癌SiHa细胞、C-33A细胞以及裸鼠肿瘤模型中,过表达FTO可部分抵消放疗辐射和顺铂对于肿瘤细胞增殖的抑制作用。首先,FTO通过去除β-链蛋白(β-catenin)mRNA的m6A修饰而增加其稳定性,并提高其蛋白表达量,随后又通过β-catenin调节了下游底物切除修复交叉互补组1(excision repair crosscomplementation group 1,ERCC1)蛋白从而影响宫颈鳞状细胞癌放化疗抵抗性,形成了FTO/β-catenin/ERCC1轴[38]。有研究发现,FTO抑制剂MA2在宫颈鳞状细胞癌中能发挥抗癌效应,使β-catenin mRNA的m6A修饰水平增高,并最终提高宫颈鳞状细胞癌对放化疗的敏感性[38]。该报道为FTO对肿瘤进展的影响提供了新的研究方向,在其他肿瘤中FTO是否存在类似的机制值得进一步探讨。
1.6 FTO与肺癌
肺癌是全世界最为常见的恶性肿瘤,其中85%的患者的组织学分型为非小细胞肺癌,而该型患者的预后常较差[73]。FTO被发现可通过调节其底物mRNA(即下文中提及的USP7)的m6A修饰而影响肿瘤的进展。LI等[39]研究发现,FTO在非小细胞肺癌组织中表达量明显增高,继而促进泛素蛋白特异性肽酶7(ubiquitin specific peptidase 7,USP7)mRNA的去m6A修饰,并增加USP7 mRNA的稳定性从而上调USP7基因的表达水平,最终促进了非小细胞肺癌的增殖。
随后,有另一项研究同样证实了FTO在非小细胞肺癌中的促癌效应。通过对比高表达野生型FTO(FTO-WT)和FTO功能缺失型突变体R96Q(FTO-R96Q)的肺腺癌A594细胞,发现仅有高表达野生型FTO的细胞出现了增殖加快和侵袭能力增强的表型。而后,通过RNA测序分析,在高表达FTO的细胞中发现了45种由于m6A修饰水平改变而表达上调的基因。功能富集分析表明大多数基因与肺癌的进展相关,其参与了细胞黏附、细胞迁移以及细胞连接等行为的调节。而当敲除FTO基因后,细胞增殖、侵袭、迁移和集落形成均被抑制,这进一步证明了FTO在肺腺癌进展中的关键作用[74]。
1.7 FTO与乳腺癌
FTO曾被认为通过参与p53基因突变和PI3K/AKT信号通路等方式影响乳腺癌的发生和进展,而在FTO被发掘出m6A“擦除器”的身份后,有关FTO与乳腺癌发生的机制又有了新的进展[75-76]。NIU[40]等研究发现,促凋亡基因Bcl-2相互作用蛋白3(Bcl-2 interacting protein 3,BNIP3)的转录物是去甲基化酶FTO蛋白介导的m6A修饰调节的下游靶点。FTO能够去除BNIP3 mRNA的3’端-非翻译区(3’-untranslated region,3’-UTR)区域的m6A修饰,从而抑制BNIP3 mRNA降解,最终导致乳腺癌细胞的凋亡减弱。因此,FTO可以通过调节BNIP3 mRNA的m6A修饰水平进而参与乳腺癌的进展。
有关FTO与乳腺癌发生和进展关系的报道不胜枚举。然而,因人种、体型和样本量等差异的影响,使得到的结果常会产生差异甚至相反的结论。此外,由于乳腺癌的发生和进展涉及多种基因的互相调控,FTO在其中所扮演的角色仍需进一步深入研究。
1.8 FTO与其他恶性肿瘤
FTO对其他恶性肿瘤的影响也已有多篇文献报道,包括肾癌、头颈鳞状细胞癌以及大肠癌的癌前病变结直肠腺瘤等[77-79]。这些报道仅通过统计学分析证明了FTO与肿瘤发生的相关性,而对其具体的作用机制尚未作出明确阐述。
FTO的3种单核苷酸多态位点(rs17817449 G、rs9939609 A和rs8050136 A)被认为与结直肠腺瘤的发生呈负相关[77]。而在头颈鳞状细胞癌组织中,FTO被发现明显高表达,却并未发现其与预后的相关性[78]。在肾透明细胞癌中,WEN等[79]根据癌组织样本中FTO的表达量从低到高将所有样本分为4组(下四分位组、第二分位组、第三分位组和上四分位组)。该研究发现,以下四分位数为界限,在界限以上FTO的表达量与肿瘤的恶性程度呈负相关,而界限以下FTO的表达量与肿瘤的恶性程度呈正相关。FTO在这些肿瘤中的机制仍需进一步阐明。
2 ALKBH5与肿瘤
2.1 ALKBH5与乳腺癌
THALHAMMER等[61]研究认为缺氧环境会诱导ALKBH5基因的表达上调。2019年诺贝尔生理学奖获得者,约翰霍普金斯大学医学院Gregg院士团队的研究同样支持该观点,并认为在乳腺癌细胞中,特异性的肿瘤缺氧微环境可以引起缺氧诱导因子HIF-1α和HIF-2α产生,从而激活ALKBH5基因的转录和翻译。ALKBH5蛋白随后发挥m6A“擦除器”作用,去除其底物NANOG同源框(nanong homeobox,NANOG)mRNA上的m6A修饰从而增加其表达的稳定性,上调NANOG蛋白的表达水平;而NANOG基因能够编码多能因子,即保持细胞多潜能性和自我更新的核心蛋白,从而调节乳腺癌干细胞的定向分化和维持。由此提示HIFs/ALKBH5/NANOG轴在乳腺癌的发生和进展过程中发挥了巨大影响[41]。
仅7个月后,Gregg院士团队就对此通路进行了补充报道[62]。该研究认为,在缺氧环境下,HIFs/ALKBH5轴激活了多种促进乳腺癌干细胞维持和富集的多能因子,除NANOG蛋白外,还包括KLF4和性别决定相关基因簇2(sex determining region Y-box 2,SOX2)蛋白。此外,还发现HIF-1α和HIF-2α的产生可以激活锌指蛋白217(zinc finger protein 217,ZNF217)基因,其编码的蛋白可以抑制m6A修饰的“书写器”METTL3的作用,与ALKBH5蛋白协同作用,以降低细胞内几种多能因子mRNA的m6A修饰水平。此外有研究报道ZNF217可以直接与NANOG和SOX2基因结合,同时在转录和转录后水平调节其表达量[62]。肿瘤干细胞在肿瘤进展中有着至关重要的作用,同时也决定着对于抗癌药物的敏感性,这些研究成果对未来乳腺癌的治疗研究有着深刻的启示作用。
2.2 ALKBH5与胶质母细胞瘤
与乳腺癌类似,ALKBH5在胶质母细胞瘤中通过调节下游底物的m6A修饰而发挥了促癌作用。ALKBH5可通过去除叉头盒转录基因M1(forkhead box M1,FOXM1)mRNA的m6A修饰,进而提高其表达量,促进GSCs的增殖,从而介导胶质母细胞瘤的进展。此外,FOXM1基因的反义链所编码的转录产物——反义链长链非编码RNA(long non-coding RNA,lncRNA)FOXM1-AS,能够极大地促进ALKBH5与FOXM1 mRNA的相互作用。下调ALKBH5基因或者FOXM1-AS基因的表达后均可有效抑制GSCs的增殖,这为胶质母细胞瘤提供了新的可能的治疗靶点[42]。
如前文所述,在乳腺癌中,缺氧环境可以诱导HIF-1α或HIF-2α,并激活ZNF217基因的转录和翻译。随后,ZNF217蛋白抑制m6A“书写器”METTL3与m6A“擦除器”ALKBH5蛋白协同作用,调低下游mRNA的m6A修饰,进而促进肿瘤干细胞增殖。在胶质母细胞瘤中,ZNF217基因同样被发现高表达,并被HIF-1α和HIF-2α诱导,参与GSCs的维持[80]。由于ALKBH5在GSCs中高表达,并与胶质母细胞瘤患者的不良预后相关,因此认为和乳腺癌相似的ALKBH5、ZNF217协同机制在胶质母细胞瘤进展中可能同样存在[42]。
2.3 ALKBH5与宫颈癌
生长阻滞特异性转录因子5(growth arrestspecial transcript 5,GAS5)基因被认为是一种抑癌基因,其可以抑制肿瘤的增殖、侵袭、转移乃至放疗抵抗性[81-82]。在子宫颈癌中,GAS5低表达被证明与患者的不良预后相关,其在宫颈癌中的表达受到其反义RNA——GAS5-AS1的调控[43,83]。在宫颈癌组织中,GAS5-AS1同样被发现低表达,并且可以抑制宫颈癌细胞的增殖和转移。类似于lncRNA FOXM1-AS在胶质母细胞瘤中的作用机制,GAS5-AS1可通过依赖ALKBH5的方式调节抑癌基因GAS的m6A修饰,高表达GAS5-AS1可去除GAS mRNA上的m6A修饰,从而减少YTHDF2阅读蛋白通过识别m6A修饰而介导的GAS mRNA降解,最终提高GAS基因的表达水平,实现对子宫颈癌进展的抑制[42-43]。
2.4 ALKBH5与胰腺癌
LncRNA KCNK15-AS1被发现在胰腺癌组织中低表达,其表达水平上调后可抑制癌组织的迁移和侵袭。同时,KCNK15-AS1的稳定性被证明与m6A的修饰水平呈负相关。荧光共聚焦显微镜显示KCNK15-AS1在胰腺癌细胞中与m6A信号共定位。将胰腺癌细胞中低表达的ALKBH5基因上调表达后,ALKBH5蛋白可以去除KCNK15-AS1的m6A修饰从而上调其表达,进而抑制上皮-间质转化(epithelial-mesenchymal transition,EMT),并阻碍了胰腺癌细胞的侵袭与转移[44]。
此外,ALKBH5还可通过去除Wnt抑制因子1(Wnt inhibitory factor 1,WIF-1)mRNA上的m6A修饰增高其表达,并抑制了Wnt信号通路。Wnt通路被阻断后,其下游基因C-Myc、细胞周期相关蛋白D1(cyclin D1)、基质金属蛋白酶2(matrix metalloproteinase-2,MMP-2)和MMP-9的表达量降低。ALKBH5在体内和体外实验中均得到证实,通过该机制不仅抑制了胰腺癌的增殖和远处转移,还提高了胰腺癌对于化学治疗的敏感性[45]。
在一项回顾性研究中,CHO等[84]的研究报道证实了ALKBH5在胰腺癌中为抑癌基因的角色。高表达水平的ALKBH5被发现与较高的总体生存率(overall survival,OS)相关,并可作为胰腺癌患者的预后预测因子,且准确性高于以TNM分期进行预测。这些研究成果为晚期胰腺癌的治疗和预后评价提供了新的研究方向。
2.5 ALKBH5与胃癌
与FTO相同,m6A去甲基化酶ALKBH5也被认为在胃癌中发挥着促癌效应[36]。ALKBH5被发现与lncRNA核副斑纹组装转录基因1(nuclearparaspeckle assembly transcript 1,NEAT1)结合,并去除其m6A修饰而调高其表达水平。高表达的lncRNA NEAT1可促进zeste 2多梳抑制复合物2亚基增强子(enhancer of zeste 2 polycomb repressive complex 2 subunit,EZH2)基因的表达上调,随后协同发挥促癌效应,最终导致胃癌细胞的侵袭和转移加速[46]。然而,学界对于ALKBH5与胃癌的关系仍有不同观点,SU等[71]通过对肿瘤基因组计划(The Cancer Genome Atlas,TCGA)数据库中的胃癌大数据进行分析后,发现较高的ALKBH5表达与胃癌患者较高的OS率相关,年龄较大和TNM分期较高的高危组中患者的胃癌组织中ALKBH5表达反而较低。
因此猜测,以上2项研究结果的差异,可能是由于样本量和采用的胃癌组织亚型不同所致,因此ALKBH5在胃癌发生中的作用与具体分子机制尚待进一步研究。
2.6 ALKBH5与非小细胞肺癌
ALKBH5在非小细胞肺癌中被认为是一种促癌基因,通过去除底物mRNA的m6A修饰来影响非小细胞肺癌进展。然而,不同于其他肿瘤的是,ALKBH5与组织金属蛋白酶抑制因子3(TIMP metallopeptidase jnhibitor 3,TIMP3)mRNA的3’-UTR区域结合并去除m6A修饰后,导致了TIMP3 mRNA的稳定性降低而非增加,使TIMP3表达水平下调。TIMP3基因表达下调后,可促进非小细胞肺癌细胞的增殖而抑制其凋亡。过高的ALKBH5和过低的TIMP3基因表达均被证实与非小细胞肺癌患者的不良预后相关[47]。
JIN等[48]研究认为,ALKBH5在非小细胞肺癌中扮演着抑癌基因的角色,并对其作用机制进行了深入的探索。研究显示,Yes相关蛋白(Yes-associated protein,YAP)可以促进非小细胞肺癌的增殖和转移,而ALKBH5则通过调节YAP mRNA表达量和抑制YAP蛋白激活2种方式抑制非小细胞肺癌进展。首先,YTHDF3蛋白是识别YAP mRNA上m6A修饰的关键分子,YTHDF1与YTHDF2蛋白均需要与YTHDF3蛋白竞争性结合从而发挥作用。在正常组织中,较多的YTHDF2蛋白与YTHDF3结合可促进YAP mRNA降解,而在非小细胞肺癌组织中,较多的YTHDF1蛋白与YTHDF3结合可促进YAP mRNA翻译。ALKBH5可以去除YAP mRNA上的m6A修饰从而抑制YTHDF1蛋白的作用,使YAP mRNA翻译受阻,最终阻碍非小细胞肺癌进展。另外,ALKBH5还可通过人抗原R(human antigen R,HuR)依赖的方式,抑制微RNA(microRNA,miR)-107表达,降低其对大肿瘤抑制激 酶2(large tumor suppressor kinase 2,LATS2)基因表达的影响,从而促进LATS2蛋白对YAP蛋白的磷酸化并抑制其活性,由此形成了一条ALKBH5/HuR/miR-107/LATS2/YAP轴[48]。
2.7 ALKBH5与骨肉瘤
LncRNA人浆细胞瘤转化迁移基因1(plasmacytoma variant translocation 1,PVT1)曾被报道在骨肉瘤中可促进肿瘤的进展[85]。ALKBH5在骨肉瘤中可去除lncRNA PVT1上的m6A修饰,导致lncRNA PVT1与m6A“阅读器”YTHDF2蛋白结合减少,从而抑制了YTHDF2蛋白介导的lncRNA PVT1降解作用,并增高了lncRNA PVT1的表达,最终促进骨肉瘤的进展。然而,在已敲除PVT1基因表达的骨肉瘤细胞中,ALKBH5的促癌作用并未失效[49]。该结果提示,ALKBH5的促癌机制并不仅限于此,可能有其他分子与信号通路参与了此过程,ALKBH5在骨肉瘤中的作用尚待进一步深入研究。
2.8 ALKBH5与卵巢上皮癌
细胞自噬被认为是一种抑癌途径,可以及时清除衰老或受损的蛋白质和细胞器,并阻止细胞癌性转化[86]。ALKBH5可通过抑制卵巢上皮癌的细胞自噬而发挥促癌作用。首先,与非小细胞肺癌类似,ALKBH5与HuR相互作用,使其表达上升,进而抑制了miR-7,导致miR-7的靶基因表皮生长因子受体(epidermal growth factor receptor,EGFR)mRNA表达水平上升,并激活经典通路EGFR/PI3K/AKT/哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR),从而抑制细胞自噬并促进细胞增殖。另外,过高的ALKBH5表达可使B淋巴细胞瘤-2(B-cell lymphoma-2,Bcl-2)基因的表达量增高,并促进Bcl-2与自噬效应蛋白Beclin 1的结合,形成“自噬开关”复合物,以抑制细胞自噬[50]。这2种机制的发现,为卵巢上皮癌提供了可能的新治疗靶点。
2.9 ALKBH5与膀胱癌
m6A修饰被发现在调控膀胱癌进展过程中发挥了巨大影响。整合素α6(integrin alpha 6,ITGA6)曾经被发现在多种恶性肿瘤中调节肿瘤细胞的EMT,而在膀胱癌中,ITGA6基因的高表达与疾病进展相关。ITGA6 mRNA是METTL3和ALKBH5的下游靶标,其m6A修饰水平受METTL3和ALKBH5的动态调节。而YTHDF1/YTHDF3能够特异性识别ITGA6 mRNA上的m6A修饰,并促进其翻译。因此,高表达ALKBH5可以抑制ITGA6 mRNA的翻译水平,减少其促癌效应,以抑制膀胱癌的进展[51]。
2.10 ALKBH5与其他肿瘤
ALKBH5与其他多种肿瘤的关系也有报道,这些肿瘤包括急性髓系白血病、肾透明细胞癌以及结肠癌[63,87-88]。
在急性髓系白血病中,ALKBH5基因的拷贝数减少,并且与TP53基因突变及急性髓系白血病的不良预后相关[63]。
在肾透明细胞癌中,可观察到ALKBH5的低表达,并发现其与肾切除术后的不良预后相关,ALKBH5低表达组患者的OS期和肿瘤特异生存期均较短[87]。
ALKBH5在结肠癌中也被发现低表达,并且与结肠癌的远处转移相关。同时,ALKBH5被发现可以作为结肠癌患者的OS率和无病生存率的独立预后影响因子。高表达ALKBH5在体外实验中可以抑制肿瘤细胞的侵袭,而在体内实验中则可抑制肿瘤的转移[88]。
3 FTO和ALKBH5抑制剂的研究进展
总结现有对于m6A去甲基化酶的研究进展发现,FTO和ALKBH5在绝大多数恶性肿瘤中均通过影响底物m6A修饰水平来激活多种信号通路以促进肿瘤进展。因而,抑制去甲基化酶FTO和ALKBH5成为多种肿瘤潜在的治疗策略,高效且特异性的FTO和ALKBH5抑制剂具有广阔的研究前景。
3.1 FTO抑制剂
由于FTO早前被认为与肥胖和能量代谢性疾病息息相关,且是首个被发现的m6A修饰“擦除器”,使得有关FTO抑制剂的研究一直是近年来的研究热点。
由于FTO属于Fe2+和2-OG依赖型双加氧酶类,所以通用型抑制剂N-草酰甘氨酸(N-oxalylglycine,NOG)可以作为抑制FTO的一种选择[89],其与2-OG有类似结构,可以竞争性抑制FTO与下游底物的结合。然而NOG较低的特异性和选择性,难以使其在某些因FTO高表达导致疾病进展的肿瘤中作为靶向治疗药物。如前所述,FTO表达改变所致的底物m6A修饰水平变化在多种肿瘤的发生和进展中有着关键作用,并且在绝大多数肿瘤中表现出了促癌效应,因此研发高选择性和特异性的FTO抑制剂迫在眉睫。
2012年,上海药物研究所以FTO晶体结构为基础,通过计算机高通量虚拟筛选和生物分析研究,报道了首个特异性较高的FTO抑制剂,即大黄酸[90]。不同于通用型抑制剂NOG,大黄酸不通过2-OG类似结构来竞争性抑制FTO,也不是一种结合2-OG辅因子的铁离子螯合剂,而是特异地竞争性结合FTO活性位点而抑制其去甲基化酶功能。大黄酸的抑制活性不仅在体外实验中通过限制性核酸内切酶消化实验和液相色谱分析等方法得到了验证,在细胞实验中,大黄酸也被证明能够调节人神经母细胞瘤细胞BE(2)-C中总mRNA的m6A修饰水平。并且,值得肯定的是,大黄酸应用于BE(2)-C细胞后,并未被发现有明显的细胞毒性,这为日后大黄酸在细胞实验研究和临床应用中增加了更多的可能性。大黄酸的报道为后期FTO高特异性抑制剂的研究提供了可靠的基础,活性位点的发现对于后期更高选择性的抑制剂探究有着重要意义,为肿瘤分子机制和相关药物开发中的表观遗传学修饰研究提供了良好的工具。
2015年,上海药物研究所在前期研究的基础上,报道了一种特异性更高的FTO抑制剂——MA2[91]。MA2是一种非甾体类抗炎药物,其通过对现存药物的高通量筛选而获得,是一种脂氧合酶环氧合酶抑制剂。有趣的是,同样在2014年报道的一篇文献中,另一种FTO抑制剂N-[3,4-二羟基-5-(4-氯苯基)-2-呋喃基]乙烷磺酰胺同样属于环氧合酶/脂氧合酶的双重抑制剂[92]。由此猜测,这类抑制剂中可能含有能够结合FTO活性位点的共同特殊结构。与大黄酸不同的是,MA2有着更高的特异性和选择性,虽然FTO与ALKBH5同属ALKB家族,并有着高度保守的共同结构,其对FTO的选择性仍远高于ALKBH5,可通过与m6A底物竞争性结合而抑制FTO效应。在细胞实验中,MA2被证明可以通过调控FTO活性进而影响人宫颈癌细胞HeLa细胞中m6A丰度,同时该调节中的m6A丰度改变被证明与另一种去甲基化酶ALKBH5无关,只与FTO的活性和表达水平相关。MA2抑制剂在该研究中的MA2/FTO复合物结构无疑可以为后期高特异性抑制剂的研究提供更多思路,以降低FTO抑制剂的脱靶效应。另外,在该研究中提出的新的FTO结合位点对后期高特异性抑制剂研究也有着积极意义。在胶质母细胞瘤和宫颈鳞状细胞癌的研究中,MA2被报道可通过抑制FTO发挥抑制肿瘤细胞增殖和提高肿瘤放化疗敏感性的作用,由此表现出了其广阔的临床应用前景[34,38]。
随后,新加坡国立大学的TOH等[93]报道了一种特异性更高的FTO抑制剂——化合物12:(2E)-4-[N’-(4-苄基吡啶-3-羰基)-肼基]-4-氧代丁-2-烯酸。在进行筛选的数十种化合物中,第12号化合物展现出了对于FTO最高的特异性和抑制效率。Glu234被发现是一个特异性更高的FTO活性位点,化合物12通过对该位点的特异性结合导致其对FTO的亲和性远高于其他ALKB家族成员,并是其他ALKB家族成员(包括ALKBH5)的30~130倍。另外,化合物12还可通过螯合金属离子的方式以增强对FTO的抑制效应。同样,化合物12的抑制效率也在细胞实验中得到了验证,在人宫颈癌细胞HeLa细胞中,化合物12已被证明可以很好地抑制FTO的m6A去甲基化酶活性。新的高特异性活性位点的发现和细胞实验的验证,无疑为后期抑制剂的研究和临床应用打下了坚实基础。
此外,除了这些高通量筛选的特异性FTO抑制剂以外,曾经被认为促癌代谢物的R-2HG已经在体内实验中被证明是一种FTO抑制剂[33]。R-2HG拥有与酮戊二酸类似的结构,因而可竞争性抑制多种Fe2+和2-OG依赖的双加氧酶,其中就包括了FTO。此外,有多种FTO抑制剂在今年被报道,包括具有抗惊厥作用的复合物7d、可以通过抑制FTO而影响小鼠糖脂代谢的恩他卡朋以及天然产物根赤壳菌素等[92,94-95]。
众多FTO抑制剂被不断报道,然而其中大部分化合物仍只经过生化或细胞实验验证。即使经过了小鼠实验体内验证的部分化合物,在差别巨大的人体内环境中,其抑制效率和安全性维持仍未可知。高特异性FTO抑制剂的开发不仅有助于肿瘤研究中甲基化修饰功能的探究,同时对于肿瘤临床治疗有也着积极意义。
3.2 ALKBH5抑制剂
不同于FTO,ALKBH5直至2013年才初次被报道具有m6A去甲基化酶作用,而有关ALKBH5抑制剂的研究鲜有报道。通过计算机软件经蛋白质组学筛选,化合物MV1035被认为是ALKBH5的抑制剂。经胶质母细胞瘤U87-MG细胞验证,MV1035不仅抑制了ALKBH5的去甲基化酶效应,并且可以减缓U87-MG细胞的迁移和侵袭[96]。XU等[60]也研究报道了柠檬酸盐是一种抑制效应较弱的ALKBH5温和型抑制剂,同时初步分析了ALKBH5结合底物的特异性环状结构,为ALKBH5的高特异性抑制剂研究奠定了基础。
总而言之,ALKBH5抑制剂的研究正处于起步阶段,受制于ALKB家族的保守结构,近年来有关高特异性ALKBH5抑制剂的报道仍极为罕见。考虑到ALKBH5作为目前已知的除FTO外唯一的m6A去甲基化酶,其可通过影响m6A修饰水平参与调节底物RNA的稳定性、翻译效率以及选择性剪接等,从而促进了包括胶质母细胞瘤在内的多种肿瘤进展,因此高效且特异的ALKBH5抑制剂亟待开发,这对于肿瘤临床治疗的重要性不言而喻。
4 结语和展望
综上所述,根据现有的文献报道,m6A去甲基化酶FTO和ALKBH5在大多数恶性肿瘤中发挥了促癌效应,仅在极少部分恶性肿瘤中抑制肿瘤进展,因而FTO和ALKBH5抑制剂的应用前景十分广阔。当然,在部分亚型恶性肿瘤的治疗中,FTO和ALKBH5激动剂也有待开发。
FTO和ALKBH5与恶性肿瘤的关系在近几年已成为研究热潮,但其具体机制仍未完全阐明。首先,尽管FTO和ALKBH5都被证实有m6A去甲基化酶作用,然而其底物选择及分布位置等均存在差异,这些差异目前尚不十分明确。其次,FTO和ALKBH5的生物学效应通常需由下游特异性靶mRNA和不同m6A阅读蛋白同时决定,对于何种阅读蛋白会结合m6A“擦除器”的靶mRNA尚待进一步鉴定。
在不同类型肿瘤中,m6A去甲基化酶对下游靶mRNA的选择差异可对肿瘤的发生和进展产生多方面影响:(1)肿瘤干细胞维持,在胶质母细胞瘤和乳腺癌中,ALKBH5通过去除靶mRNA FOXM1和NANOG上m6A修饰来促进肿瘤干细胞的维持[41-42];(2)肿瘤细胞增殖,FTO去除C-Myc mRNA m6A修饰可促进白血病细胞和胰腺癌细胞的增殖[33,45];(3)肿瘤细胞侵袭和转移,在膀胱癌和胰腺癌中,ALKBH5分别通过调节靶mRNA和靶lncRNA的m6A修饰水平进而影响肿瘤细胞EMT,干预肿瘤的侵袭和转移[45,51]。
去甲基化酶的生物学效应也同时取决于不同类型的阅读蛋白。以经典的阅读蛋白YTH家族为例,YTHDF2蛋白被发现可识别大部分mRNA上的m6A修饰位点,促进mRNA向降解场所转移[23]。如YTHDF2可识别斑马鱼胚胎中母源mRNA从而加速其清除,而在急性髓系白血病中,YTHDF2识别了m6A修饰后介导了C-Myc mRNA的衰退,并抑制了肿瘤细胞的增殖[33,97]。YTHDF1被报道在与m6A位点结合后,常与翻译起始因子eIF3相互作用,从而促进mRNA翻译[25]。如YTHDF1与溶酶体蛋白酶mRNA上的m6A位点结合,可促进其翻译,并导致抗原被降解,进而影响树突状细胞的抗原提呈能力,最终抑制肿瘤免疫应答[98]。
总而言之,若要探讨m6A去甲基化酶与肿瘤发生和进展的关系,在鉴定下游靶mRNA的同时,进行相关阅读蛋白的鉴别或可使分子机制的研究事半功倍。
在总结本综述所引用的文献时发现,m6A“擦除器”在不同恶性肿瘤中存在如下几种共同机制:(1)在大多数恶性肿瘤中FTO和ALKBH5通过去除靶mRNA或靶lncRNA的m6A修饰,从而改变其稳定性,进而发挥生物学效应[34,38-39,42,46-47,49,64,85,88];(2)在非小细胞肺癌及卵巢上皮癌中,ALKBH5可以依赖HuR的方式调控微RNA(microRNA,miRNA,miR),通过抑制microRNA的靶mRNA进而发挥作用[48,87];(3)m6A“擦除器”在恶性肿瘤中可影响多种经典癌基因或信号通路,包括C-Myc和p53基因以及Wnt、EGFR/PI3K/AKT/mTOR通路等[33,35,37,45,75-76,87]。
综上所述,FTO和ALKBH5在多种恶性肿瘤进展中的关键作用毋庸置疑,但是其如何调控各种蛋白和下游信号通路的机理尚未完全阐明。结合m6A对RNA出核、翻译效率和可变剪接等多方面的影响,推断FTO和ALKBH5在恶性肿瘤中的调控机制所涉及的蛋白和通路可能远超现有文献报道。相信在不久的将来,FTO和ALKBH5会成为多种恶性肿瘤诊断和预后的标志物乃至关键治疗靶点。
猜你喜欢
保健医苑(2022年6期)2022-07-08
现代仪器与医疗(2021年4期)2021-11-05
广东农业科学(2017年10期)2018-01-25
天津医药(2016年9期)2016-10-20
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10
上海工运(2015年11期)2015-08-21
疑难病杂志(2014年12期)2014-04-16
中国中医药现代远程教育(2014年22期)2014-03-01
遗传(2014年3期)2014-02-28
河南科技(2014年22期)2014-02-27
