
NADH 脱氢酶1 基因变异致Leigh 病1 例报告并文献复习
2021-05-01 11:21赵东敬汤继宏张兵兵王曼丽
临床儿科杂志 2021年4期
赵东敬 汤继宏 刘 影 张兵兵 肖 潇 王曼丽
苏州大学附属儿童医院 1.神经内科,2.儿科研究所(江苏苏州 215025)
Leigh病是儿童和婴幼儿常见的神经退行性疾病,主要累及基底神经节、间脑、脑干、脊髓[1],其临床表现多样,主要为精神运动发育落后或倒退、生长发育落后以及乳酸性酸中毒等[2]。Leigh病病因复杂,其发病机制主要是线粒体呼吸链障碍导致ATP生成减少,从而引起中枢神经系统进行性变性[3]。国内外研究证实,线粒体呼吸链5种酶复合物(Ⅰ、Ⅱ、Ⅲ、Ⅳ和Ⅴ)、丙酮酸脱氢酶复合物缺陷以及线粒体转运RNA 变异等均可引起Leigh 病[4-6]。因此相关位点基因变异,均有可能为Leigh 病的发病原因。现报告1 例NADH脱氢酶1(NADH:Ubiquinone Oxidoreductase Core Subunit 1,ND1)基因新发现的变异m.3688G>A导致Leigh 病,国内未见该ND 1 中m.3688 G>A 基因变异报道。
1 临床资料
患儿,男,1 岁3 个月,因表情淡漠2 个月,于2018 年10 月收入苏州大学附属儿童医院神经内科。患儿出生史及母亲孕期无特殊。患儿4 个月会抬头、1岁会爬,入院时仍独坐不稳,不能独站及扶走。患儿入院前2 个月,头部外伤后出现表情淡漠,当时无哭闹、无呕吐,家长未予重视,后症状持续未见好转。外院头颅CT示双侧额叶、侧脑室旁、基底节区、中脑及丘脑区多发低及稍低密度灶;双侧额颞部脑沟增宽。入院体格检查:神志清,精神稍差,表情淡漠,检查欠合作,颈软,呼吸平稳,双肺呼吸音粗,未及干湿啰音;心律齐;未及杂音;腹软,未及包块,肝脾肋下未及;四肢肌力对称,无明显肢体瘫痪表现,肌张力稍低下,膝反射可引出,病理征阴性。实验室检查:血乳酸7.6 mmol/L,血常规、血氨、生化检查均未见异常,甲状腺功能未见异常,血尿遗传代谢筛查未见异常。头颅磁共振成像(MRI)示双侧基底节、丘脑、大脑脚可见多发小斑片状长T 1、长T 2 信号影,FLAIR 和DWI上病灶均呈高信号影,病灶大致呈对称性分布,双侧额顶叶皮质可见脑回样长T 1 长T 2 及FLAIR 高信号影(图1)。入院时脑电图检查示双侧大脑半球基本波变慢,未见典型尖波、棘波及其复合波发放。
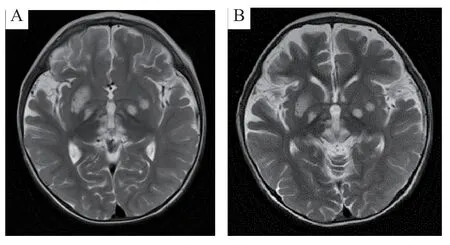
图1 患儿头颅MRI T2WI 表现
入院后考虑为急性播散性脑脊髓炎,予免疫治疗。静脉滴注丙种球蛋白(2 g/kg,分3天用完),大剂量甲基泼尼松龙(15 mg/kg,静脉滴注,连续3 天)冲击治疗,随后甲基泼尼松龙序贯减量(每3天减半量)治疗2周,效果欠佳,患儿仍表情淡漠,精神运动发育落后无明显改善。患儿病因未明,治疗效果欠佳,经医院医学伦理审核,家属签署知情同意书后行全外显组家系项目及线粒体高敏感项目检测协助诊断。
结果显示ND 1 基因上发现了一个意义未明的变异m.3688G>A(图2),样本中的变异率为9.76%,该变异会导致ND1的第128位氨基酸由Ala变异为Thr。在Polyphen-2 网站(http://genetics.bwh.harvard.edu/ggi/pph2/df7a6a7967f13718a7f87af1b5b08d8443302 0e8/7016882.html)中预测该变异位点为可能有害(分值为1.000)”。分值高低就是判断是否有害的标准,从0到1,分值越高越有害。
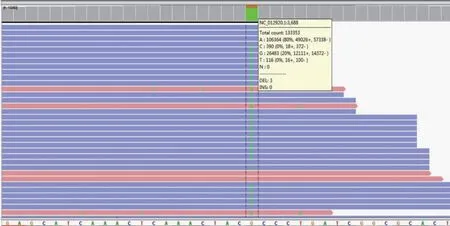
图2 ND1 上变异m.3688G>A 基因图谱
治疗2 周后,复查患儿头颅MRI 示双侧基底节、丘脑、大脑脚以及大脑皮质区可见多发异常信号影,大部分DWI为高信号,病灶大致呈对称性分布,双侧额顶叶皮质可见脑回样长TI、长T2、FLAIR稍高信号影,较入院时变化不明显。
2 讨论
Leigh病是一种遗传异质性的儿童及婴幼儿时期常见的亚急性坏死性脑病,其病因多种多样,但其神经病理学改变是一致的,即表现为脑干、脊髓、小脑等部位的对称性坏死性病变[1-2]。本例患儿主要临床表现为表情淡漠、运动发育落后,乳酸水平增高,头颅MRI表现为双侧基底节、丘脑、大脑脚以及大脑皮质区可见多发异常信号影,病灶大致呈不完全对称分布,临床表现及影像学特点均符合Leigh病[2]。
Leigh 病病因复杂,以呼吸链复合物Ⅳ即细胞色素C 氧化酶(cytochrome coxidase,COX)缺陷最多见,SURF1基因异常是导致COX缺陷的常见原因[7]。对330例Leigh病患者的mtDNA变异分析显示,96例(32%)存在mtDNA变异,其中MT-ND和MT-ATP6基因变异最为常见[2]。另外有研究发现ND 基因变异与复合体I 的分离缺陷有关,进一步证实ND 基因变异与Leigh病相关[8]。在ND4基因中还曾发现一种新的线粒体DNA 变异(T 11984 C),并推测可能有更多其他的线粒体基因变异与Leigh 病相关[9]。之后在3 例Leigh 病兄妹中发现同源的ND 1,m.3697 G>A 变异(G131S),由于三兄妹均携带同源m.3697G>A变异,且均具有Leigh病表型,因此推测ND1,m.3697G>A变异可导致Leigh病[10]。随着基因检测技术的推广和应用,与Leigh病相关的基因更多变异被发现。
MT-ND 1,也被称为线粒体编码的NADH 泛醌氧化还原酶核心亚基1,在生成NADH 脱氢酶1 中起主要作用,这是线粒体呼吸链复合物I 的一种蛋白。该基因变异引起线粒体呼吸链复合物I 功能障碍[11]。在对240 例线粒体神经肌肉病患者的研究中发现5 例有5 个新的致病变异,其中1 例的疾病表型为 Leigh 病,通过mtDNA序列分析发现其ND1基因携带同源3688 G>A 变异,该变异导致高度保守的残基(p.A 128 T)发生改变,从而导致丙氨酸变异为苏氨酸;同期发现其肌肉和成纤维细胞均存在特异性和孤立性复合物 I 缺陷[12]。孤立性复合物I 缺陷是儿童线粒体疾病最常见的生化缺陷,占20%~30%,Leigh 病的发病与复合物 I 缺陷密切相关[13]。本例患儿的线粒体基因也为ND 1,3688 G>A 变异,结合临床表型,考虑为Leigh 病。虽然ND 1,3688 G>A 基因变异尚未收录至线粒体致病性变异(统一由Mitomap数据库收录,已收录 83 个致病变异),但3688 G>A基因变异可能是Leigh病的一种新发现的致病基因。
大多数致病性mtDNA 变异是异质性的,每个细胞内都有不同数量的变异mtDNA。表型表达最终取决于变异mtDNA的比例或脆弱组织中野生型mtDNA的数量。只有当变异数量超过临界阈值水平时,才会出现呼吸链的生化缺陷[14]。在对3 168份新生儿脐带血样本中10个线粒体点变异的频率分析中发现,以匹配的母血样估计新生变异率,其中15 个子代检测到mtDNA变异,这15例阳性患儿mtDNA变异率均值为43%[15]。本例患儿的样本变异率为79.76%,其母亲为无症状者。当个体从携带低水平变异mtDNA 且无症状的母亲那里遗传到高比例的变异mtDNA 时,线粒体疾病的家族才首次出现临床症状[16]。本例患儿的母亲无症状,而患儿发病,从遗传学角度来说不一定会首先考虑线粒体遗传方式,故很可能忽略对mtDNA进行测序。但是本例患儿临床上疑似线粒体疾病,经mtDNA 测序,最终发现可能致病的变异。提示即使遗传方式看起来不符合线粒体遗传方式,但是临床表型上可能为线粒体病时,要积极对mtDNA 进行测序分析。
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02
体育科技文献通报(2022年3期)2022-05-23
中草药(2022年5期)2022-03-03
中国生物化学与分子生物学报(2022年1期)2022-02-26
中国典型病例大全(2022年1期)2022-01-10
实用肿瘤学杂志(2021年3期)2021-11-29
智慧健康(2021年33期)2021-03-16
中外医疗(2018年12期)2018-09-03
中西医结合心血管病电子杂志(2016年23期)2017-03-03
中国实用医药(2016年30期)2016-12-28
