
溃疡性结肠炎患者及其配偶的肠道菌群构成与多样性分析
2021-04-25 10:21吴佳倩马芳笑吴佩钟继红刘英超
浙江临床医学 2021年3期
吴佳倩 马芳笑 吴佩 钟继红 刘英超*
溃疡性结肠炎(UC)主要是以累及直肠和结肠的慢性非特异性炎症性肠病。研究表明,UC在欧洲的发病率已达0.97~57.9/10万人,亚洲和中东近几年发病率快速上升,达0.15~6.5/10万人[1-2]。目前认为UC是免疫、遗传、环境及肠道菌群等多因素共同作用的结果。研究表明,某些肠道菌群释放大量毒素损伤黏膜,诱导细胞死亡并刺激细胞炎症因子释放,引起肠道黏膜炎症反应[3-4]。此外,外界环境同样是引起UC发病的因素之一[5]。本文比较UC患者与其同一生活环境下患者家属,以及与正常体检人群肠道菌群的差异性,探讨生活环境与肠道菌群环境间的相关性在UC发病中的作用。
1 资料与方法
1.1 临床资料 选取2019年1月至7月浙江中医药大学附属第二医院UC患者8例(UC组)及其配偶8例(UF组),同时招募本院体检健康者8例(ZC组)。本项目经本院医学伦理委员会批准。(1)纳入标准:①年龄18~60岁,性别不限;②根据患者临床表现、肠镜及病理结果,参照《炎症性肠病诊断与治疗的共识意见(2018年)》[6]的UC诊断标准,由消化内科及病理科医师共同确诊为UC。③UC配偶组需与UC患者共同生活至少5年、无全身系统性疾病且肠镜检查正常;④健康人群组需与UC患者无血缘关系且不在同一家庭环境下生活;⑤健康人群组无全身系统性疾病且肠镜检查未见肠道病变;⑥所有入选者均签署知情同意书。(2)排除标准:①重度或急性暴发型UC患者;②近2个月内使用抗生素、益生菌制剂及清洁灌肠者;③既往有消化道疾病手术史;④妊娠及哺乳期女性、儿童及精神病患者;⑤患有严重心、脑、肺、肝、肾疾病或高血压、心脏病、糖尿病等慢性疾病。
1.2 方法 (1)粪便样本采集:采集所有入选者自然排出的新鲜粪便(>2 g),迅速将粪便装于无菌样品管中,样品管冰浴1 h内运送至中心实验室无菌条件下进行分装,液氮冷却后置于-80 ℃超低温冰箱中保存。(2)样本DNA提取、基因组PCR扩增及16S rRNA测序:应用QIAamp DNA Stool Extraction Kit试剂盒从粪便样本中提取肠道微生物基因组DNA后,使用引物341F(5'-CCTACGGGNGGCWGCAG-3')和805R(5'-GACTACHVGGGTATCTAATCC-3')[7]对细菌16S rRNA基因的V3-4可变区进行PCR扩增[7]。扩增条件为95℃解链3 min,95℃30 s,55℃,30 s,72℃45 s,25个循环,72 ℃延伸5 min。用1.0%琼脂糖凝胶电泳对PCR产物进行质检并纯化。利用Nextera XT Index Kit中的引物在待测DNA的两端接入adaptor序列,用于后续MiSeq上机测序。(3)测序数据分析:剔除原始序列中不含引物、长度在300~480 bp以外、质量评分≤20、含有模糊碱基N及嵌合体的序列,得到最终用于分析的优质序列。将相似度在97%的优质序列聚类为一个操作分类单元(OTU),并用SILVA数据库进行生物学鉴定。最终得到所有OTU表,采用Qiime 软件进行多样性指数分析、主成分分析及菌落组成分析。
1.3 统计学方法 采用SPSS 26.0统计软件。计量资料以(±s)表示,多组间比较用方差分析,多重比较采用LSD-t检验;方差不齐时,采用Kruskal-wallis H检验。计数资料组间比较采用卡方检验。P<0.05为差异有统计学意义。
2 结果
2.1 三组基线资料比较 见表1。
表1 三组基线资料比较(±s)

表1 三组基线资料比较(±s)
组别 n 年龄(岁) 男/女(n) BMI(kg/m2)ZC组 8 36.00±7.31 4/4 20.70±1.99 UC组 8 43.38±11.75 4/4 21.09±0.68 UF组 8 41.89±18.39 4/4 21.11±1.32
2.2 肠道菌群物种多样性分析 物种多样性分析采用Alpha多样性指标,Alpha多样性是反映丰富度和均匀度的综合指标。丰富度即种类数目;多样性即群落中个体分配上的均匀性。本研究中群落丰富度的指数以Chao1指数表示。群落多样性的指数以Shannon指数表示。(1)Chao1指数:每条曲线代表一个样本,末端数字为实际测序条数。Chao1指数越大,表明群落的丰富度越高。测序序列从15,000起曲线趋于平坦,表明此时测序已趋于饱和,测序数据量合理。三组间Chao1指数差异无统计学意义(P>0.05)。见图1。(2)Shannon指数:每条曲线代表一个样本,末端数字为实际测序条数。Shannon指数值越高,表明群落的多样性越高。测序序列曲线较早趋于平坦,表明此时测序已趋于饱和,测序数据量合理。三组间Shannon指数差异无统计学意义(P>0.05)。见图2。

图1 Chao1指数曲线

图2 Shannon指数曲线
2.3 肠道菌群门分类水平分析 所有样本共检出18个细菌门(见图3)。其中,厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)、变形杆菌门(Proteobacteria)、梭杆菌门(Fusobacteria)以及放线菌门(Actinobacteria)的相对丰度依次递减,且总量占所有菌群数量的99.9%。与UC组比较,ZC组及UF组中厚壁菌门与梭杆菌门的相对丰度平均值均增加,厚壁菌门分别增加5.73%和3.74%,梭杆菌门分别增加1.06%和6.09%;而拟杆菌门的相对丰度均值分别减少2.83%和10.87%。与ZC组比较,UC组及UF组中变形菌门的相对丰度均值分别增加2.77%和3.28%;放线菌门分别增加1.21%和1.62%。三组样本间肠道菌群的相对丰度差异无统计学意义(P>0.05)。见表2。

图3 门分类水平上三组样本肠道菌群的相对丰度
表2 三组肠道菌群门水平的相对丰度比较[%,(±s)]

表2 三组肠道菌群门水平的相对丰度比较[%,(±s)]
组别 厚壁菌门 拟杆菌门 变形杆菌门 梭杆菌门 放线菌门ZC组 53.90±23.64 41.60±22.38 2.31±1.66 1.08±2.64 1.01±0.95 UC组 48.17±12.43 44.43±11.24 5.08±4.63 0.02±0.017 2.22±1.99 UF组 51.91±1.09 33.56±14.40 5.59±4.33 6.11±10.19 2.63±5.26
2.4 肠道菌群属分类水平分析 所有样本共检出240个菌属,相对丰度前35的菌属在各组样本中的构成情况见图4。ZC组优势菌属:厚壁菌门的粪杆菌属(Faecalibacterium)、拟杆菌门的拟杆菌属(Bacteroides)及普雷沃菌属(Prevotella);UC组及UF组优势菌属:拟杆菌门的拟杆菌属、 厚壁菌门的粪杆菌属及罗斯菌属(Roseburia)。所有菌属中共有15个菌属在属分类水平上组间分布差异有统计学意义,其中前5类菌属差异有显著统计学意义。与UC组比较,ZC组及UF组的考拉杆菌属(Phascolarctobacterium)的相对丰度增高(P<0.05);与UF组比较,ZC组及UC组的锥形杆菌属(Pyramidobacter)的相对丰度降低(P<0.05);与ZC组比较,UC组及UF组奥尔森菌属(Olsenella)、霍尔德曼菌属(Holdemania)、克雷伯菌属(Klebsiella)的相对丰度增高(P<0.05)。(见图5及表3)。

图4 属分类水平上三组样本肠道菌群的相对丰度
表3 三组肠道菌群属水平相对丰度比较[%,(±s)]

表3 三组肠道菌群属水平相对丰度比较[%,(±s)]
注:与UC组比较,*P<0.05;与UF组比较,#P<0.05;与ZC组比较,△P<0.05
组别 考拉杆菌属 奥尔森菌属 锥形杆菌属 霍尔德曼菌属 克雷伯菌属ZC组 1.58±0.75*0.0005±0.001 0.00±0.00# 6.64±4.83 0.02±0.03 UC组 0.21±0.52 0.006±0.007△0.00±0.00# 8.59±8.47△1.74±3.24△UF组 2.23±2.86*0.006±0.016△0.004±0.005 8.00±7.52△1.04±1.74△
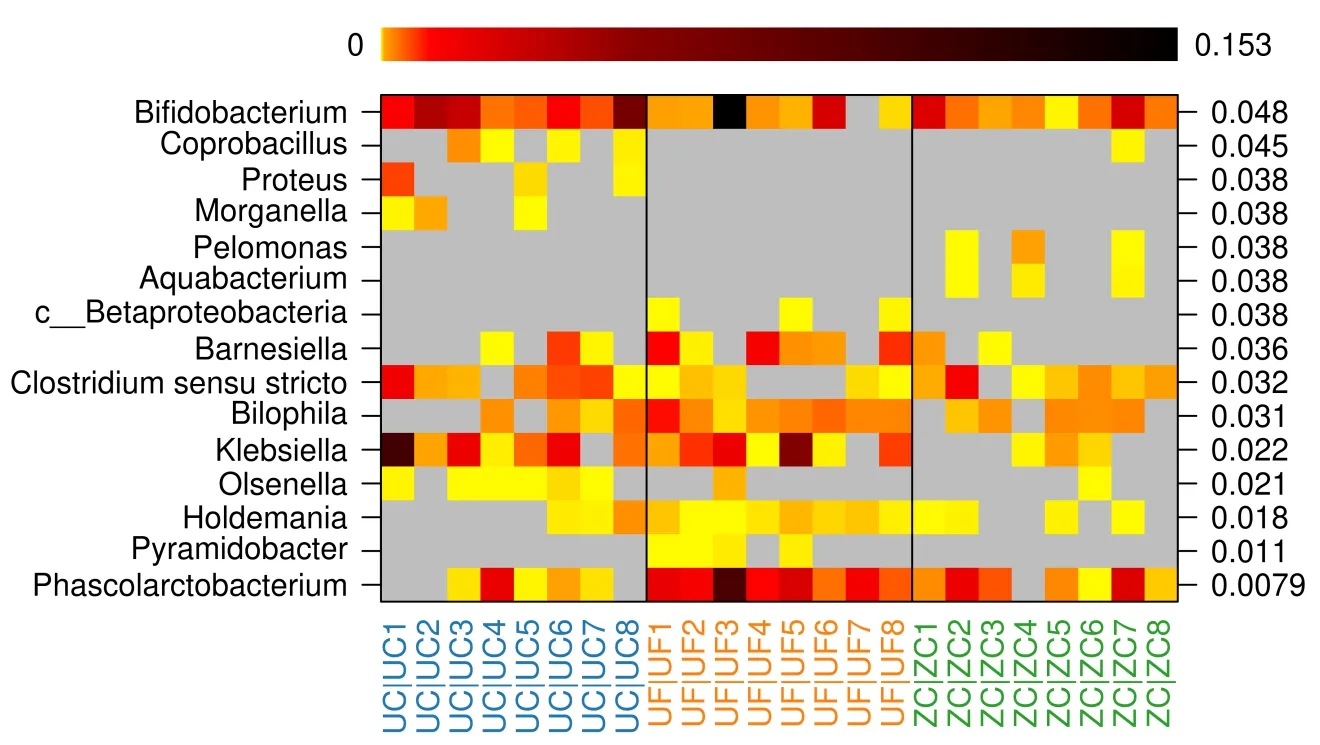
图5 属分类水平上三组样本差异性肠道菌群的相对丰度Image图
3 讨论
肠道菌群与溃疡性结肠炎的发病机制密切相关,在正常生理状态下,肠腔内有益菌群及有害菌群相互制约、相互依存,共同维持肠道稳态。一旦肠道微生态失衡,有益菌减少而致病菌繁殖增加,侵袭、破坏肠黏膜屏障进而引发肠黏膜免疫反应,导致慢性炎症和组织损伤[8-10]从而引起UC发病。
本研究显示,三组间Chao1指数及Shannon指数的差异无统计意义。表明三组间菌群丰富度及群落物种多样性存在相似性,但每个菌种具体相对丰度的差异还需进一步的分析。比较健康人群、UC患者及其配偶的差异性菌属,发现UC患者肠道内考拉杆菌属的相对丰度明显降低。考拉杆菌属是短链脂肪酸——丁酸及丙酸的生产者,后者可经SCFA / GPCR途径抑制NLRP3炎症小体激活及炎症因子分泌;同时诱导结肠调节T细胞(Treg)的分化,维持结肠免疫耐受及应答间的平衡[11-12]。LUKAS等[13]研究发现,考拉杆菌属在UC肠道中明显减少。本研究结果与上述研究相一致,提示考拉杆菌属可能是UC患者发病的标志性菌群。
此外,UC组及UF组优势菌属均为拟杆菌门的拟杆菌属、 厚壁菌门的粪杆菌属及罗斯菌属,而健康人群组的优势菌属为厚壁菌门的粪杆菌属、拟杆菌门的拟杆菌属及普雷沃菌属。与ZC组比较,UC患者组及其配偶组肠道内奥尔森菌属、霍尔德曼菌属及克雷伯菌属的相对丰度升高。奥尔森菌属归属于放线菌门,是一种条件性致病菌[13]。研究报道奥尔森菌属的减少有助于减轻肠黏膜炎症损伤[14]。霍尔德曼菌属归属于厚壁菌门,有研究表明饮酒会引起此菌属丰度的上升[15],其可能参与一些疾病的发生发展过程。克雷伯菌属归属于变形杆菌门,是一种寄居机体肠道的条件致病菌。机体健康状态下保持沉默,但当机体抵抗力下降、肠道菌群失调时,其将迅速繁殖、破坏肠黏膜屏障,从而引发遗传易感人群严重的肠道炎症反应[16]。本研究已排除血缘关系等其他混杂因素后,发现UC组与UF组肠道内霍尔德曼菌属、克雷伯菌属及奥尔森菌属的相对丰度较健康人群显著升高。因此,作者推测以上2种菌属可能随着生活环境或日常饮食改变而变化,在一定条件下可能促发UC易感人群发病。相比传统细菌培养鉴定,16S rRNA基因测序技术对菌群构成的分类准确率更高。本研究从UC患者及其配偶肠道中检测到可能与UC发生发展相关的差异菌群:如考拉杆菌属、奥尔森菌属及克雷伯菌属。上述菌群的发现将为针对肠道菌群靶点的UC精准治疗提供新思路。
综上所述,考拉杆菌属、奥尔森菌属及克雷伯菌属可作为UC肠道菌群失调预后及治疗的潜在生物学标志物。此外,人体肠道菌群结构与数量因生活环境的不同会产生一定差异,但这种差异不一定会引起机体发病,多种因素共同作用导致UC易感人群的肠道微环境失衡是UC的重要发病机制。
猜你喜欢
科学技术与工程(2022年26期)2022-11-01
河南医学研究(2022年19期)2022-10-19
中国农学通报(2022年14期)2022-06-01
分子诊断与治疗杂志(2022年4期)2022-05-30
野生动物学报(2022年2期)2022-05-16
雪豆月读·低年级(2021年1期)2021-09-10
雪豆月读·低年级(2021年2期)2021-03-22
国际消化病杂志(2021年1期)2021-03-05
湖北农业科学(2019年22期)2019-12-23
快乐语文(2018年7期)2018-05-25
