
不同施肥模式对茶园土壤细菌多样性的影响
2019-12-23 01:22毛迎新黄丹娟王红娟谭荣荣陈勋王友平
湖北农业科学 2019年22期
毛迎新 黄丹娟 王红娟 谭荣荣 陈勋 王友平

摘要:以菜子餅+茶叶配方肥(YSJF1)、脲甲醛缓释肥(YSJF2)、茶叶配方肥(YSJF3)、畜禽粪商品有机肥+茶叶配方肥(YSJF4)、菜子饼+脲甲醛缓释肥(YSJF5)、包膜控释肥(YSJF6)、茶叶复混肥(YSJF7)、不施肥(YSJF8)和习惯施肥(YSJF9)处理后的茶园土壤为研究对象,利用Ion S5TMXL高通量测序技术,以16S rRNA基因为靶标,研究不同施肥模式对茶园土壤细菌数量、多样性和群落结构的影响,分析细菌数量、多样性与土壤理化性质的关联性。在门分类水平上,27个样品共鉴定获得46个类群,其中变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)、放线菌门(Actinobacteria)、绿弯菌门(Chloroflexi)、厚壁菌门(Firmicutes)、奇古菌门(Thaumarchaeota)、拟杆菌门(Bacteroidetes)为优势类群,相对丰度占87.08%~96.76%。不同施肥处理间变形菌门、酸杆菌门、放线菌门、绿弯菌门的相对丰度差异显著。不同施肥处理的ACE指数、Chao1指数、Shannon指数和Simpson指数分别为656.34~962.36、655.79~1 040.41、5.22~6.54和0.925 0~0.969 3。相关性分析和冗余分析结果表明,土壤pH、有机质含量和碱解氮与细菌丰度、α多样性指数和细菌群落结构有一定的相关性,土壤pH是影响细菌群落重要的环境因子。
关键词:茶园;施肥模式;细菌菌落多样性;16S rRNA
中图分类号:S154.3 文献标识码:A
文章编号:0439-8114(2019)22-0065-06
DOI:10.14088/j.cnki.issn0439-8114.2019.22.015 开放科学(资源服务)标识码(OSID):
Effects of different fertilization managements on tea garden soil bacterial diversity
MAO Ying-xina,HUANG Dan-juana,WANG Hong-juana,TAN Rong-ronga,CHEN Xuna,WANG You-pingb
(a.Institute of Fruit and Tea;b.Institute of Plant Protection and Soil Fertilizers/Key Laboratory of Integrated Pest Management Crops in Central China,Ministry of Agriculture,Hubei Academy of Agricultural Sciences,Wuhan 430064,China)
Abstract: A field trial was conducted in tea plantation, nine different soil samples were collected, including rapeseed cake fertilizer and tea formula fertilizer(YSJF1), urea-formaldehyde slow released fertilizer(YSJF2), tea formula fertilizer(YSJF3), animal manure and tea formula fertilizer(YSJF4), rapeseed cake fertilizer and urea-formaldehyde slow released fertilizer(YSJF5), coated controlled release fertilizer(YSJF6), tea compound fertilizer(YSJF7),no fertilization(YSJF8), and the chemical fertilizer(YSJF9). Using Ion S5TMXL high-throughput sequencing technology, targeting the 16S rRNA gene, the effects of different fertilization patterns on the number, diversity and community structure of soil bacteria in tea plantation, and the correlation between the number, diversity and soil physical and chemical properties was analyzed. At the phylum level, forty-six phyla were obtained in all the treatments, among which Proteobacteria, Acidobacteria, Actinobacteria, Chloroflexi, Firmicutes, Thaumarchaeota and Bacteroidetes were the common dominant bacteria, accounting for 87.08%~96.76% of the total reads. There were significant differences in relative abundance of Proteobacteria, Acidobacteria, Actinobacteria and Chloroflexibetween among different treatments. The index of Ace, Chao1, Shannon and Simpson were 656.34~962.36, 655.79~1 040.41, 5.22~6.54 and 0.925 0~0.969 3, respectively. Correlation analysis and redundancy analysis showed that soil pH, organic matter content and alkali-hydrolyzed nitrogen were correlated with bacterial abundance, alpha diversity index and bacterial community structure, and soil pH was an important environmental factor affecting bacterial community.
利用Uparse软件(Uparse v7.0.1001)[10]对所有样品的全部有效数据进行聚类,以97%的一致性(Identity)将序列聚类成为操作分类单元OTUs(Operational Taxonomic Units),筛选OTUs中出现频数最高的序列作为OTUs的代表序列。
用Mothur方法与SILVA132(http://www.arb-silva.de/)[11]的SSUrRNA数据库[12]进行物种注释分析(设定阈值为0.8~1.0),获得分类学信息并分别在各个分类水平统计各样本的群落组成。使用MUSCLE[13](Version 3.8.31,http://www.drive5.com/muscle/)软件进行快速多序列比对,得到所有OTUs序列的系统发生关系。最后对各样品的数据进行均一化处理,以样品中数据量最少的为标准进行均一化处理,后续的Alpha多样性分析和Beta多样性分析都是基于均一化处理后的数据。
1.4 数据处理
利用SPSS 22.0对土壤理化性质、细菌16S rRNA相对丰度、多样性指数等数据进行多重比较、方差分析和相关性分析;环境因子对细菌群落结构影响的冗余分析采用R3.3.2软件完成。
2 结果与分析
2.1 土壤样品测序结果
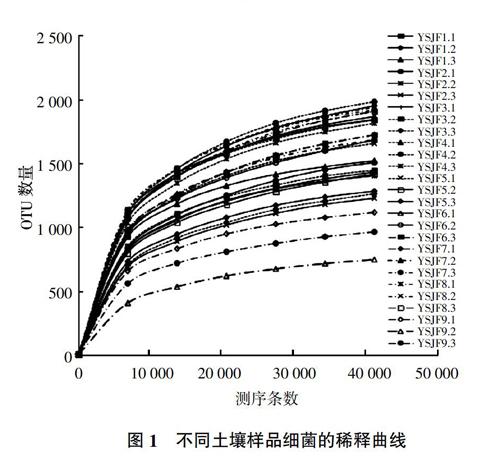
利用Ion S5TMXL平台对细菌16S rRNA基因的V3-V4区测序,27个土壤细菌样品通过对Reads剪切过滤,平均每样品测得83 166条Reads,经过质控平均得到78 142条有效数据,平均长度为407.93 bp,质控有效率达94.01%。以97%的一致性(Identity)将序列聚类成为OTU(Operational taxonomic units),共得到4 173个OTU。样品OTU稀释曲线趋向平坦,说明测序数据量合理(图1)。
2.2 不同施肥模式对土壤细菌群落组成的影响
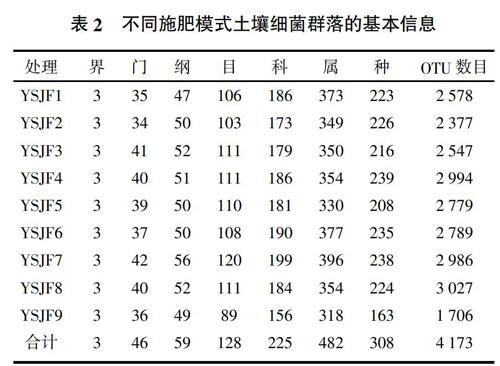
在97%相似性阈值下,9个处理(YSJF1至YSJF9)分别聚类为2 578、2 377、2 547、2 994、2 779、2 789、2 986、3 027和1 706个细菌OTU(表2)。不同处理间共有的OTU数目为789个,代表性的物种分别属于γ-变形菌纲(Gammaproteobacteria)、酸杆菌纲(Acidobacteriia)、α-变形菌纲(Alphaproteobacteria)、Nitrososphaeria、酸微菌纲(Acidimicrobiia)、纤线杆菌纲(Ktedonobacteria)、拟杆菌纲(Bacteroidia)等。不施肥处理不含有其独有的细菌OTU。与不施肥处理相比,YSJF1、YSJF6施肥处理均增加了纤维杆菌纲(Fibrobacteria)、甲烷微菌纲(Methanomicrobia)等细菌物种;YSJF2施肥处理增加了Rhodothermia等物种;YSJF3施肥处理增加了其独有未归类的螺旋体纲的细菌(unidentified_Spirochaetes);YSJF4施肥处理增加了互营养菌(Synergistia)等菌群;YSJF5施肥处理增加了未归类的泉古菌门的细菌unidentified_Crenarchaeota等;YSJF7施肥处理增加了甲烷杆菌纲(Methanobacteria)、Chitinivibrionia、unidentified_Zixibacteria等独有的细菌物种;YSJF9处理增加了互营养菌、Rhodothermia等物种;同时施肥处理也存在细菌物种的减少,如习惯施肥處理减少了全噬菌纲(Holophagae)、未归类的古细菌unidentified_Archaea等6个细菌物种。
2.3 不同施肥模式对土壤细菌相对丰度的影响
在门分类水平上,共鉴定获得46个类群,其中,变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)、放线菌门(Actinobacteria)、绿弯菌门(Chloroflexi)、厚壁菌门(Firmicutes)、奇古菌门(Thaumarchaeota)、拟杆菌门(Bacteroidetes)为优势类群,相对丰度分别为30.45%~61.59%、5.85%~27.42%、2.34%~23.40%、0.74%~14.48%、0.11%~14.41%和0.93%~13.62%,相对丰度共占87.08%~96.76%(图2)。
不同施肥模式间变形菌门(P<0.05)、酸杆菌门(P<0.01)、放线菌门(P<0.01)、绿弯菌门(P<0.001)的相对丰度差异显著。调查发现,YSJF1处理组放线菌门高于其他处理组,绿弯菌门显著高于其他处理组,拟杆菌门则低于其他处理组。YSJF9处理组变形菌门显著高于其他处理组,但其放线菌门、绿弯菌门、奇古菌门和芽单胞菌门则低于其他处理组。
在纲分类水平上,共鉴定获得59个纲,γ-变形菌纲、酸杆菌纲、α-变形菌纲、Nitrososphaeria、酸微菌纲、纤线杆菌纲、拟杆菌纲为主要优势类群,相对丰度分别为15.14%~52.96%、5.57%~24.75%、8.54%~23.20%、0.11%~14.16%、1.16%~13.59%和0.39%~12.32%,相对丰度共占63.68%~80.10%(图3)。
不同施肥模式间嗜热油菌纲(P<0.05)、酸杆菌纲(P<0.01)、γ-变形菌纲(P<0.001)、酸微菌纲(P<0.001)、纤线杆菌纲(P<0.001)的相对丰度差异显著。YSJF1处理组酸微菌纲、梭菌纲(Clostridia)均高于其他处理组,纤线杆菌纲显著高于其他处理组,拟杆菌纲则低于其他处理组。YSJF9处理组拟杆菌纲高于其他处理组,γ-变形菌纲显著高于其他处理组,但其酸微菌纲、纤线杆菌纲和嗜热油菌纲则低于其他处理组(图3)。
[7] 顾松松,胡秋龙,刘仲华,等.不同类型茶园土壤细菌多样性及群落结构研究[J].茶叶通讯,2019,46(2):162-169.
[8] LANGILLE M G I,ZANEVELD J,CAPORASO J G,et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences[J].Nature biotechnology,2013,31(9):814-821.
[9] MARTIN M. Cutadapt removes adapter sequences from high-throughput sequencing reads[J].Embnet journal,2011,17(1):10-12.
[10] HAAS B J,GEVERS D,EARL A M,et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons[J].Genome research,2011,21(3):494-504.
[11] EDGAR R C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads[J].Nature methods,2013,10(10):996-998.
[12] WANG Q,GARRITY G M,TIEDJE J M,et al. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J].Applied and environmental microbiology,2007,73(16):5261-5267.
[13] QUAST C,PRUESSE E,YILMAZ P,et al. The SILVA ribosomal RNA gene database project:Improved data processing and web-based tools[J].Nucl Acids Res,2013(41):590-596.
[14] 陳声明,张立钦.微生物学研究技术[M].北京:科学出版社,2006.209.
[15] JANSSEN P H. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes[J].Applied and environmental microbiology,2006,72(3):1719-1728.
[16] YOU J,DAS A,DOLAN E M,et al. Ammonia-oxidizing archaea involved in nitrogen removal[J].Water research,2009,43(7):1801-1809.
[17] GRIFFITHS R I,THOMSON B C,JAMES P,et al. The bacterial biogeography of British soils[J].Environmental microbiology,2011,13(6):1642-1654.
[18] ZHALNINA K,DIAS R,QUADROS P D DE,et al. Soil pH determines microbial diversity and composition in the park grass experiment[J].Mol Ecol,2015,69(2):395-406.
[19] 王伏伟,王晓波,李金才,等.施肥及秸秆还田对砂姜黑土细菌群落的影响[J].中国生态农业学报,2015,23(10):1302-1311.
猜你喜欢
昆明医科大学学报(2022年2期)2022-03-29
中国比较医学杂志(2020年4期)2020-05-26
国际呼吸杂志(2019年22期)2019-12-09
水生生物学报(2019年4期)2019-07-20
生物安全学报(2019年3期)2019-02-15
川北医学院学报(2019年6期)2019-02-10
水生生物学报(2015年1期)2015-02-28
应用海洋学学报(2014年4期)2014-11-22
天然产物研究与开发(2014年6期)2014-04-27
河南科技(2014年18期)2014-02-27
