
A deletion mutation along with a novel DNA variation in OCRL cause Lowe syndrome in a child with multiple secondary manifestations
2021-04-17 02:42AlirezaPaniriSadeghFattahiAhmadRasoulinejadHalehAkhavanNiaki
Alireza Paniri, Sadegh Fattahi, Ahmad Rasoulinejad, Haleh Akhavan-Niaki,4
1Department of Genetics, Babol University of Medical Sciences, Babol 4717647745, Iran
2North Research Center, Pasteur Institute of Iran, Amol 4619332976, Iran
3Department of Ophthalmology, Rouhani Hospital, Babol University of Medical Sciences, Babol 4717647745, Iran
4Akhavan-Niaki Genetics Laboratory, Babol 4714957534, Iran
Dear Editor,
We report the deletion and insertion mutations of oculocerebrorenal syndrome of Lowe (OCRL) in a child who has been presenting typical symptoms of Lowe syndrome with many secondary phenotypes including dental abnormalities, bleeding problems, mild hypochromic anemia,and nystagmus in a non-Caucasian population for the first time.Lowe syndrome is a rare, X-linked recessive genetic disorder with a prevalence of 1:500 000 in population[1].Lowe is caused by a mutation in OCRL gene containing 24 exons, which encodes inositol polyphosphate 5-phosphatase protein that regulates several cellular processes including cell proliferation, cytoskeletal function, and membrane trafficking.The symptoms could be variable depending on type and location of mutation[2]. The three most common signs and symptoms of Lowe syndrome are ocular manifestations,central nervous system (CNS) disorders, and renal tubular dysfunction[3]. This condition almost exclusively affects males with severe symptoms in comparison with females which are asymptomatic, nonetheless, they might present variable phenotypes due to random X-inactivation[4]. Usually, patients are referred to the ophthalmologist due to ocular symptoms including cataracts and glaucoma in the first month of life[5].Furthermore, renal tubular dysfunction and CNS disorders often appear gradually a few years later[6].
The patient, a boy aged about 5y, was the first child of the family, and was born at 39wk of gestation with a natural delivery and weighed 3.150 kg. The informed consent was obtained from the legal guardian. Also, his height and head circumference at birth were 49 and 35 cm, respectively.Congenital bilateral cataracts were operated under several anesthesia at the age of 2 and 4mo, and aphakia was corrected with wearing glasses (OD: 23-2×180 and OS: 25-2×180). The cup to disc ratio (CDR; right and left: 0.1) and intraocular pressure (right IOP: 12 mm Hg, left IOP: 14 mm Hg) were bilaterally measured. The fundus was regularly followed after surgery and showed no abnormality. Also, nystagmus and hypotonia were seen in the first few days after birth. Moreover,psychomotor retardation has been observed in the first years of life; the patient could raise his head and hand, but couldn’t sit, stand, and walk without support. He began to spoke at 2 years old for the first time and only could say mama and baba. The auditory skills were well developed and he reacted normally to sounds around. The brain magnetic resonance imaging (MRI) performed at two years of age has presented clear cortical atrophy, compensated ventriculomegaly, and white matter abnormality (leukodystrophy). Besides, severe dental abnormality was a notable symptom in this case (Figure 1).Renal dysfunction has been confirmed with urine analysis showing proteinuria, microscopic hematuria, epithelial cells,hyaline cast, and moderately amorphous phosphate crystals.Furthermore, urine biochemistry tests have revealed an elevated calcium/creatinine ratio (about 0.49). The complete results of biochemistry analysis are shown in Table 1. The plasma calcium and phosphorus levels were in the normal range(probably due to taking calcium and phosphorus supplements)but liver function tests showed increased levels of aspartate aminotransferase (AST) while alanine aminotransferase (ALT)levels were normal. Hematological tests have demonstrated decreased levels of hemoglobin, hematocrit, mean corpuscular volume (MCV), and mean corpuscular hemoglobin (MCH)with mild hypochromic anemia (Table 2) which were corrected after taking folic acid and iron supplements. Also, he has taken vitamin D, C, A, and L-carnitine supplements since 1-year-old.Moreover, the levels of platelet were within a normal range(minimum value: 163×103μL). Thyroid hormone analysis showed normal levels of T4 (8.7 μg/dL) and TSH (2.1 mIU/L)after taking thyroid supplements. Venous blood was collected into EDTA containing tube from the child and his parents while chorionic villi were obtained from the fetus at week 12 of gestation for mutation detection. DNA extraction, PCR and Sanger sequencing were performed for patient and his parents.It is noteworthy that the mother was referred to us at week 7 of gestation without prior genetic analysis. Also, corresponding to the clinical manifestations of the child we decided to directly investigate OCRL gene responsible for Lowe syndrome. Given mother’s gestational age, we firstly sequenced those exons with higher mutation frequencies worldwide (exons 18 to 24 and their flanking intron regions). Therefore, we checked all remaining exons except exons 1 and 6.
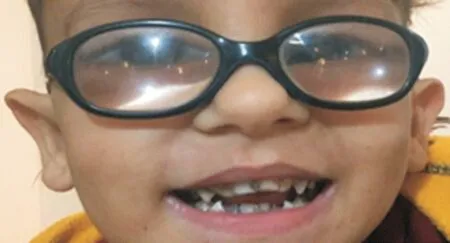
Figure 1 Upper and lower teeth abnormality.
The results of sequencing have been illustrated that the child was hemizygote for a deletion c.1925_1926delCT (p.S641fs)in exon 18 along with an insertion c.1879+39_1879+40 insCTC in intron 17 (Figure 2A and 2B). His mother was also carrier for this mutation (Figure 2C) without any symptoms nonetheless the father showed a wild type genotype. The measurements of bilateral CDR, IOP and visual activity of mother were within the normal range. Also, bilateral examination of her foveal thickness was within the normal limit (Figure 3). However, her lenses showed mild cortical opacification. She didn’t reveal any known history of abortion and currently is pregnant with a twin that one of them died at 9wk of gestation and the other one was revealed to be heterozygote female upon chorionic villus sampling and genotyping at week 12 of gestation; mother and fetus were healthy at month 6 when the manuscript was submitted.Both parents were consenting to participate to this study.Collectively, the spectrum of clinical manifestations of patients with Lowe syndrome could be very variable depending on location and type of mutation. Therefore, detecting mutations is helpful to predicting protein function and genotypephenotype correlation. Consequently, accession to the clinical details of patients is playing a key role in diagnosis of rare conditions. It’s more important when they are referred to a genetic counselor to have another baby.
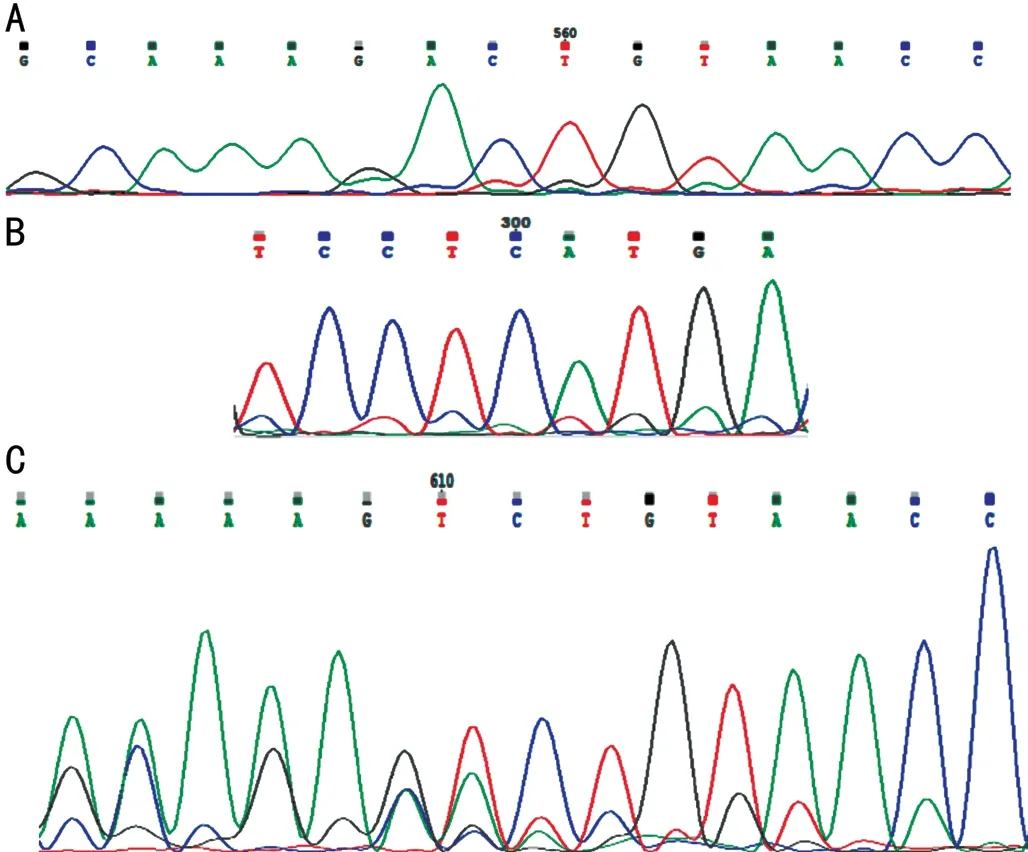
Figure 2 Electrophoregrams of exon 18 and intron 17 of OCRL after Sanger sequencing A: Sequencing result of the child shows a deletion mutation in exon 18; B: A novel insertion mutation in intron 17 of the child; C: Sequencing result of the mother shows a deletion mutation in intron 18; exon 18 was sequenced using reverse primer.
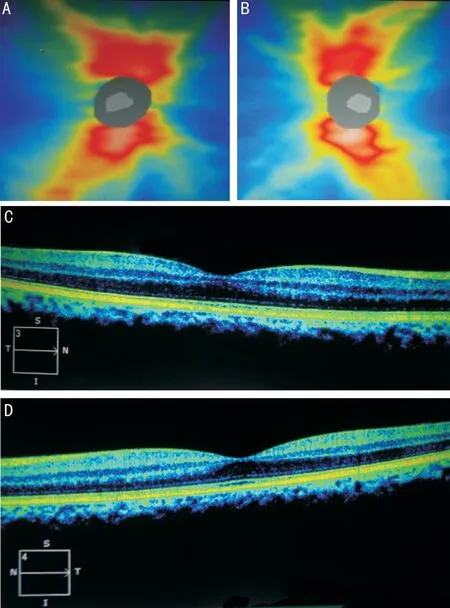
Figure 3 Bilateral measurement of the cup to disc ratio and foveal thickness of mother A: Retinal nerve fiber layer thickness of right eye; B: Retinal nerve fiber layer thickness of left eye; C: Foveal thickness of right eye; D: Foveal thickness of left eye.
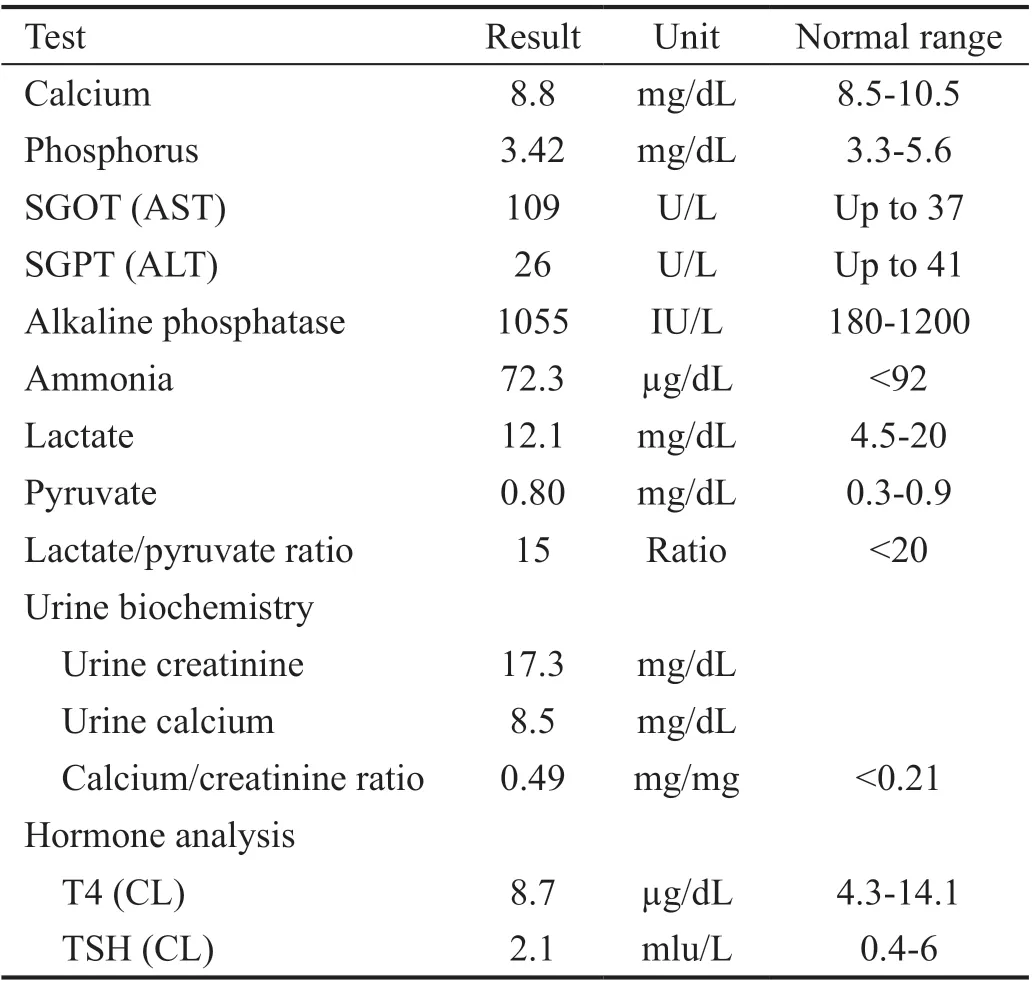
Table 1 Biochemistry, urine, and hormone analyses
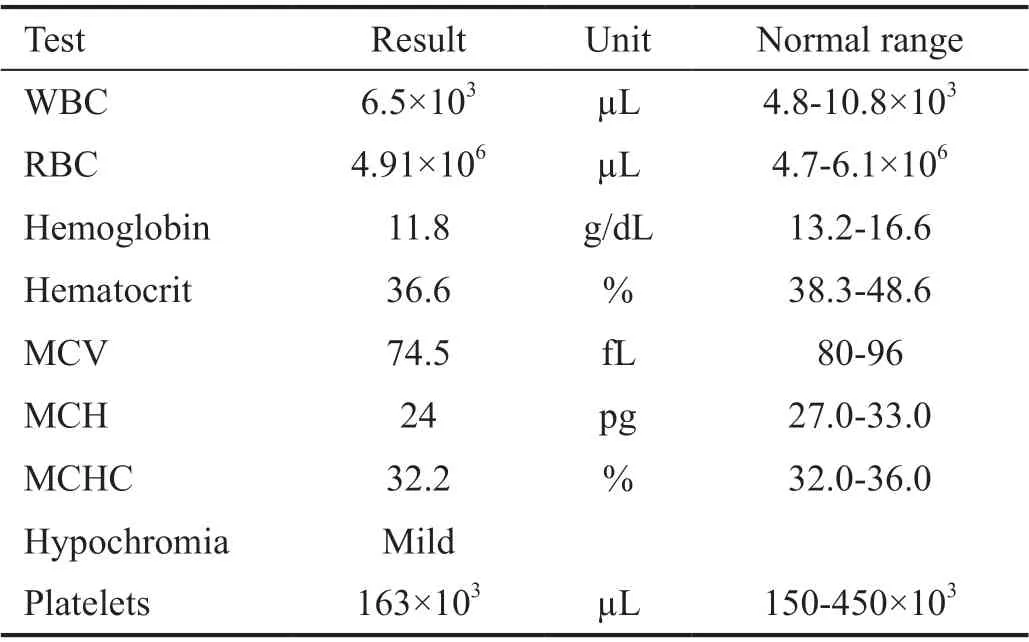
Table 2 Hematological indices analysis
ACKNOWLEDGEMENTS
Conflicts of Interest: Paniri A,None;Fattahi S,None;Rasoulinejad A,None;Akhavan-Niaki H,None.
 International Journal of Ophthalmology2021年4期
International Journal of Ophthalmology2021年4期
- International Journal of Ophthalmology的其它文章
- Prevalence and risk factors of dry eye disease in young and middle-aged office employee: a Xi’an Study
- YM155 inhibits retinal pigment epithelium cell survival through EGFR/MAPK signaling pathway
- Clinical features and treatment outcomes of intraocular lymphoma: a single-center experience in China
- Trends in research related to high myopia from 2010 to 2019: a bibliometric and knowledge mapping analysis
- A simple new technique for the induction of residual posterior vitreous cortex removal and membrane peeling
- Differential degeneration of rod/cone bipolar cells during retinal degeneration in Royal College of Surgeons rats