
ldentification of a novel FOXL2 mutation in a fourthgeneration Chinese family with blepharophimosis-ptosisepicanthus inversus syndrome
2021-04-17 02:43WeiNingRongMeiJiaoMaWeiYangShiQinYuanXunLunSheng
Wei-Ning Rong, Mei-Jiao Ma, Wei Yang, Shi-Qin Yuan, Xun-Lun Sheng
Department of Ophthalmology, Ningxia Eye Hospital,People’s Hospital of Ningxia Hui Autonomous Region (First Affiliated Hospital of Northwest University for Nationalities),Yinchuan 750002, the Ningxia Hui Autonomous Region,China
Abstract
● KEYWORDS: blepharophimosis-ptosis-epicanthus inversus syndrome; FOXL2; mutation; Chinese
INTRODUCTION
Blepharophimosis-ptosis-epicanthus inversus syndrome(BPES; OMIM# 110100) is an autosomal dominant developmental disorder with the prevalence of approximately 1 in 50 000[1]. It has been reported there were no prevalence differences in gender, race, or ethnicity[2]. The clinical features of BPES include complex eyelid/ocular deformities characterized by blepharophimosis, ptosis, epicanthus inversus and telecanthus. BPES has been divided into two types based on the presence or absence of premature ovarian failure (POF).Type I is characterized by eyelid malformation and female infertility due to POF, and type II is characterized only by eyelid malformation[1].
Up to now, six pathogenic genes (FOXL2, CRBP, INHBA,EGFR, HOX1, BPESC1) have been found to be associated with BPES and the majority (88%) of patients with BPES harbor pathogenic mutations in the FOXL2 gene[3]. FOXL2 gene is 2.7 kb and located on chromosome 3q22.3 with one exon, which encodes 376 amino acids. The FOXL2 protein contains a 110-amino-acid DNA-binding forkhead domain(residues 54-164) and a polyalanine (poly-Ala) tract of 14 residues of unknown function (residues 221-234)[4].

Figure 1 Pedigree of the family with BPES.
FOXL2 is an important factor in embryonic development of the eyelids and ovaries, as well as in the growth of the female gonads[5]. To date, more than 200 mutations on the FOXL2 gene have been reported in patients with BPES[6]. Most of the genetic defects found in BPES were caused by genetic mutations, and others were caused by genomic rearrangements involving the FOXL2 gene[7]. Although there are some studies have suggested that different mutations in FOXL2 were associated with BPES types, direct genotype-phenotype association remains to be further confirmed due to the lack of de novo genetic study and the clinical heterogeneity of BPES patients[8]. Therefore, identifying novel FOXL2 mutations and further investigating their contributions to the pathogenesis of BPES will provide biomarkers for early detection of BPES and potential implications for therapeutic intervention.
In the present study, we identified a novel mutation of the FOXL2 gene from a large Chinese family displaying typical type II BPES phenotypes.
SUBJECTS AND METHODS
Ethical ApprovalThis research confor med to the Declaration of Helsinki and was prospectively reviewed and approved by the Ethics Committee of Ningxia Eye Hospital. All participants gave written informed consent prior to enrolment.
PatientsA non-consanguineous Chinese pedigree with BPES members (Figure 1) was recruited from the Ningxia Eye Hospital. Twenty-one members of this family were recruited in this study, including 13 patients and 8 unaffected family members. Medical records of each participant were reviewed. Routine ophthalmic examinations were performed on all participants, including uncorrected visual acuities, best corrected visual acuities (BCVAs), slit-lamp examination,fundus examination, color face photography. The length and height of the eyelid fissure, the distance between inner canthus,the muscle strength of levator eyelid and the muscle strength of frontal muscle were measured. The diagnostic criteria of BPES included blepharophimosis, ptosis, epicanthus inversus and telecanthus. We extracted DNA samples from these 21 members. In addition, to determine the type of BPES, the reproductive function the female patients were examined especially. The proband (Ⅳ-34) in this family was a 10-yearold girl who presented with the typical features of BPES,including small palpebral fissure, ptosis, epicanthus inversus and telecanthus.
Targeted NGS Approach and in Silico AnalysisSamples of peripheral venous blood (5 mL) were collected from each participant for genomic DNA extraction using a QIAmp DNA Mini Blood Kit (Qiagen, Hilden, Germany). To reveal the pathogenic mutation in this family, targeted NGS approach was selectively performed on the proband (Ⅳ-34), patient (Ⅲ-32) and normal family member (Ⅳ-32). The target sequence capture chip including the reported 403 eye anterior segment disease genes, according to known disease genes and mutations,customize specific oligonucleotides nucleoside acid probe,nucleic acid sequence data reference to Ensembl database(https://www.ensembl.org) and UCSC website CCDS database(https://genome.ucsc.edu). The target genomic region was captured in liquid phase by Agilent SureSelect exon targeting sequence enrichment system of Applied BioSystems (ABI,USA). Library preparation, qualification, and NGS were further performed on the Illumina Hiseq2000 platform (Illumina, Inc.,San Diego, CA, USA). After sequencing, the raw data were saved as a FASTQ format, then followed the bioinformatics analysis, then the data would be transformed to VCF format,variants were further annotated by ANNOVAR (http://annovar.openbioinformatics.org/en/latest/) and associated with multiple databases, such as 1000 genome (http://www.1000genomes.or/), dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/),HGMD (http://www.biobase-international. com/product/hgmd), and confirmed associated pathogenic gene mutational site by PCR and Sanger sequencing. The sequences were verified by comparing them to the human genomic sequence of FOXL2 (GenBank NG_012454.1) and screened for mutations.
In Silico AnalysisThe possible effects of the amino acid changes on the function of FOXL2 protein was predicted by using the PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), the Sorting Tolerant from Intolerant (SIFT) algorithm(http://sift.jcvi.org/www/SIFT_enst_submit.html), and the Mutation Taster (http://www.mutationtaster.org/). The predicted score of polyphen2 (HDIV/HVAR) is close to 1, indicating that it is probably damaging (D), otherwise it is possibly damaging (P) or benign (B). Analyzed by Sift,amino acids with probabilities <0.05 are predicted to be deleterious (D). The scores of Mutation Taster analysis are between 0 and 1, it is more likely to be disease causing with the score closer to 1. Sanger sequencing was performed for intrafamilial cosegregation analysis. The allele frequencies of the mutations in the normal population were viewed in 1000 Genome (http://browser.1000genomes.org/index.html), EVS(http://evs.gs.washington.edu/EVS/) and EXAC (http://exac.broadinstitute.org/) databases.
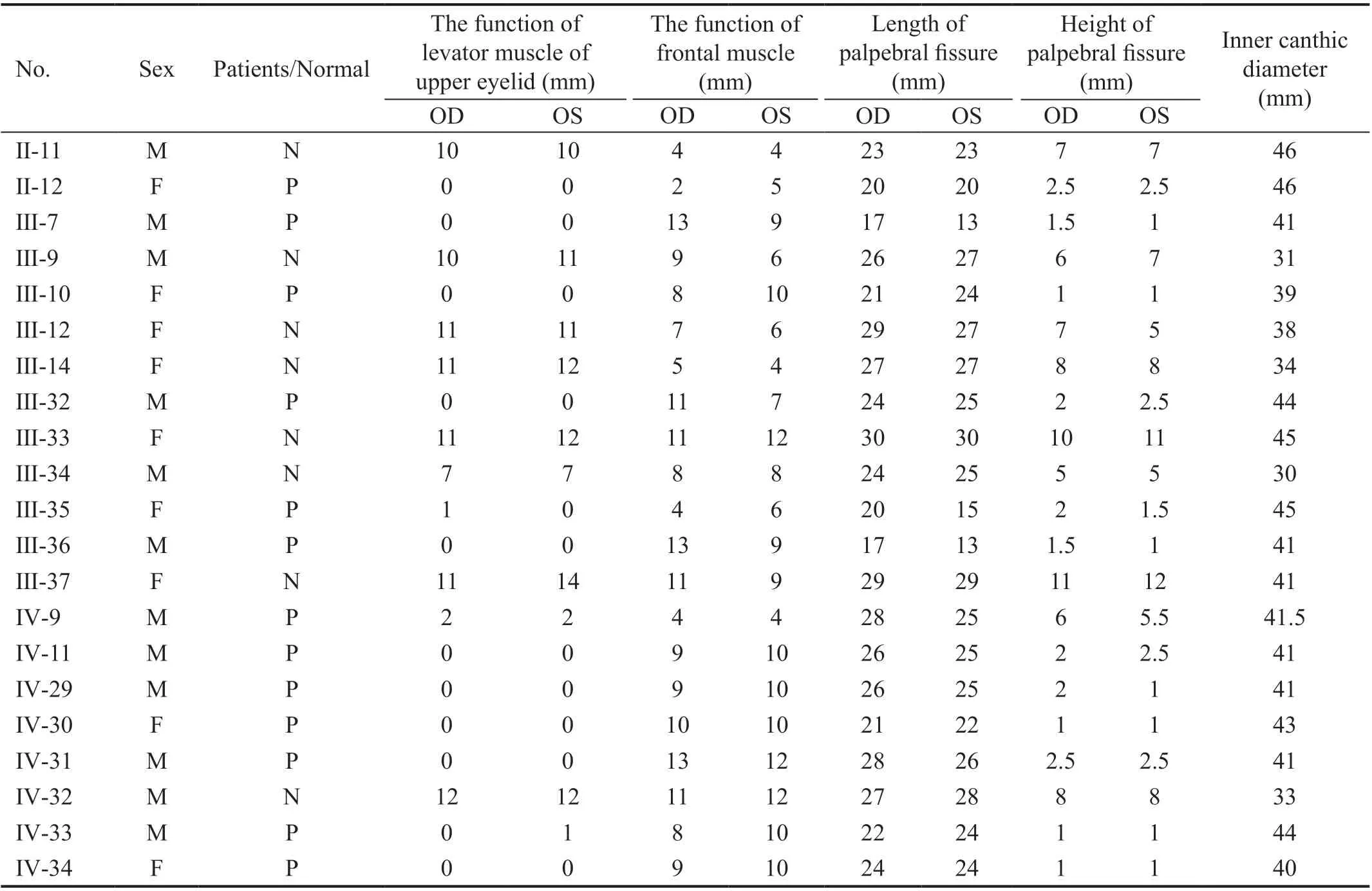
Table 1 Clinical features of the pedigree
Conservational AnalysisUNIPROT knowledgebase(https://www.uniprot.org), a central hub for the collection of functional information on proteins, with accurate, consistent and rich annotation, was used to obtain amino acid sequence of FOXL2 from different species, and then they were aligned and analyzed by alignment tool-Multalin (http://sacs.ucsf.edu/cgi-bin/multalin.py). Low change of the amino acid site we focused between these sequences indicates that it is highly conserved among species and the mutation occurring at this site is more likely to affect the structure and function of related protein.
RESULTS
Clinical FeaturesWe collected a four-generation, 104-member Chinese family with 29 individuals suffering from BPES, as shown in Figure 1. The pattern of inheritance in this family is autosomal dominant. A total of 13 BPES patients and 8 normal family members were included in our study. All patients in this family demonstrated the typical features of BPES, including small palpebral fissures, ptosis, telecanthus and epicanthus inversus. Some patients had signs of mandible lifting and head backward (Table 1). None of the patients showed microcephaly, intellectual disability or other malformations.All adult female patients had offspring (Figure 1).
The proband Ⅳ-34, a 10-year-old girl, the visual acuity was 10/20 in both eyes with a mild hyperopia. The clinical features include a flattened bridge of the nose, wide inner canthus spacing in both eyes, inverted inner canthus covering the lacrimal montanus, ptosis of the upper eyelid, and upper eyelid occlusion of the upper 2/3 pupil. The length of palpebral fissure is 24mm and the height of palpebral fissure is 3mm of both eyes, the inner canthic diameter is 42mm (40-44mm). The function of levator muscle of upper eyelid of both eyes is 0 and the function of frontal muscle of both eyes is 9 mm. Both her father and brother were diagnosed with BPES, and her mother was normal (Figure 2).
According to clinical manifestations, this family was diagnosed BPES type Ⅱ. Amblyopia was found in 8 patients (16 eyes), 7 with hyperopia, 2 with myopia, and 12 with astigmatism.
Mutation AnalysisUsing the panel sequencing we initially identified a novel heterozygous insertion mutation c.672_701ins GCGGCTGCCGCCGCAGCTGCTGCAGGCG CT (p.Ala234_Gly235linsAAAAAAAAGA) (Figure 3)of FOXL2 gene in the proband Ⅳ-34 and Ⅲ-32 and then segregated the disease status, finally confirmed in other eleven patients and absence in the unaffected members respectively by sanger sequencing (Figure 3). The heterozygous insert mutation has not been previously reported in the literature and was also not detected in the normal people database of gene sequencing company (Agilent Technologies, Inc.).The mutation was not found in ESP database, 1000 database and EXAC database (that is, the mutation frequency of this mutation in normal population was 0). This mutation resulted in the insertion of 10 amino acids into the encoded polyalanine chain, which increased the number of original polyalanine chains from 14 to 24, resulting in an extended protein.
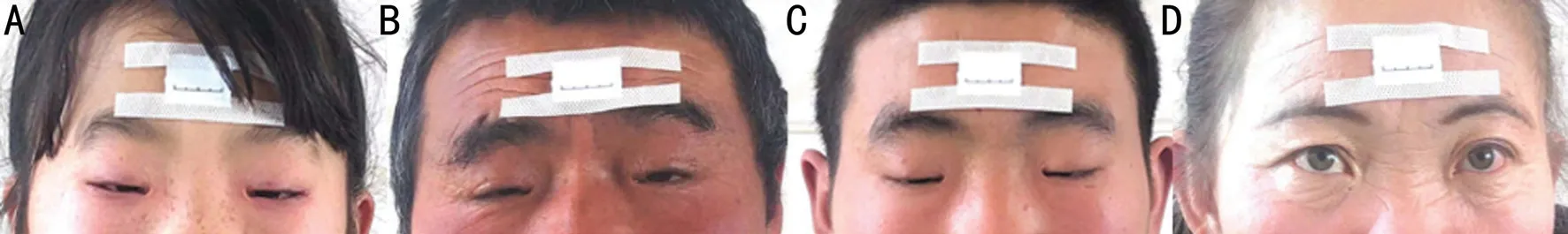
Figure 2 Photos of eye appearance in the BPES pedigree A: The proband; B: Proband’s father; C: Proband’s brother; D: Proband’s mother.
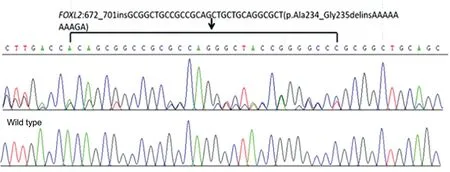
Figure 3 Sequence analysis A heterozygous insertion mutation c.672_701ins GCGGCTGCCGCCGCAGCTGCTGCAGGCGCT (p.Ala234_Gly235linsAAAAAAAAGA) of FOXL2 gene were identified in the affected individuals.

Figure 4 Conservation analysis Orthologous protein sequence alignment of FOXL2 from humans, bovine, pig (S. scrofa), and rabbit. The mutated residue 234-235 showed highly conserved among species.
Furthermore, in silico analysis by the prediction programs for variant of c.672_701ins GCGGCTGCCGCCGCAGCTGCT GCAGGCGCT, mutation taster obtained prediction score of 1 and indicated disease causing. This amino acid change was probably damaging predicted by PolyPhen-2 (score=1.000)and SIFT (score=0.00) programs.
In this Chinese BPES family, the heterozygous insertion mutation c.672_701ins GCGGCTGCCGCCGCAGCTG CTGCAGGCGCT (p.Ala234_Gly235linsAAAAAAAAGA)were identified and protein function predicted. In this mutation, the alanine at position 234 was highly conserved among different species by proteomic conservation analysis(Figure 4). In general, this pathogenic variant is likely to be the causative mutation for the disease phenotype in this family.The segregation of the p.Ala234_Gly235linsAAAAAAAAGA mutation within the family was verified because it was only present in all affected individuals but not present in other normal family members. The population frequency analysis showed this mutation in normal population was 0. There are no other potential pathogenic variants detected in this study. The well-established correlations between the genes mutated and the disease phenotypes for this family.
DISCUSSION
BPES is an autosomal dominant disorder caused by monoallelic mutations of the FOXL2 gene[9]. In this study, we reported a novel heterozygous insertion mutation c.672_701ins GCGGCTGCCGCCGCAGCTGCTGCAGGCGCT(p.Ala234_Gly235linsAAAAAAAAGA) in the FOXL2 gene in a BPES pedigree.
In our study, the clinical diagnosis was BPES type Ⅱ. As previously reported, genetic defects involving the FOXL2 gene can be identified in nearly 90% of BPES patients[10].The mutations included missense mutations, premature stop codons, expansions of the region encoding the poly-Ala tract and frameshift mutations leading to a shorter or longer protein[11]. However, the correlation between BPES types and mutation type remains to be further defined. Poly-Ala expansion mutations usually lead to BPES type II, whereas truncations before the poly-Ala tract often led to BPES type I[12-13]. The heterozygous insertion mutation detected of our research also verified the relationship between the genotype and phenotype.
FOXL2 mutations have been classified according to their predicted function of the protein. The mutations that cause truncated proteins can be classified into four categories: protein without forkhead domain (A); protein with a partial forkhead domain (B); protein with complete forkhead domain, but without poly-Ala tract (C); and predicted frameshift variants that lead to truncated proteins with complete forkhead (D) and poly-Ala domains. In addition, group E includes mutations causing elongation of FOXL2 protein, group F includes in-frame changes,group G are missense mutations, and group H comprises chromosomal rearrangements disrupting FOXL2[14-15].Thus, in our study, the insertion mutation c.672_701ins GCGGCTGCCGCCGCAGCTGCTGCAGGCGCT(p.Ala234_Gly235linsAAAAAAAAGA) was classified to group E.
Two FOXL2 hotspots have been identified in BPES subjects.One of them is located at the polyproline tract domain(amino acids 283 a 288), accounting for 13% of all described intragenic mutations. The other FOXL2 hot spot is located at the polyalanine tract and is associated with about 30%of all pathogenic FOXL2 changes[14]. The mutation of our study was located at this region and was predicted to extend the tract of alanine. Poly-Ala expansions cause aggregation,mislocutions, and intranuclear mobility of the protein[16].These suggest that the novel mutation c.672_701ins GCGGCT GCCGCCGCAGCTGCTGCAGGCGCT (p.Ala234_Gly235 linsAAAAAAAAGA) in the FOXL2 gene may be pathogenic and thus responsible for the pathogenesis of BPES patients.
It has been reported that the variation of different base changes c.672_701dupGCGGCTGCAGCCGCAGCTGCTGCAGC CGCT (p.Ala234_Gly235insAAAAAAAAAA) at the same position as the mutation in this study at the same position was detected in patients with a number of BPES patients[17-19].About 30% of the patients caused by this duplicate mutation related to type Ⅱ BPES, but rarely reported in the Asian people.In this study, the insertion mutation of this locus was reported for the first time, which again proved that this region was the hot spot of BPES mutation.
Due to congenital ptosis, inverted inner canthus and narrow eyelid fissure at birth, patients with BPES are disturbed,inhibited and deprived of visual stimulation during the critical period of visual development, which eventually leads to visual development disorders and amblyopia. Early diagnosis and treatment of congenital ptosis will help to prevent and manage these eye abnormalities. We examined the refractive status of 8 patients from this family after excluding other eye diseases.Among them, 2 adults presented severe amblyopia without surgical treatment, and 2 adults showed moderate amblyopia after surgical treatment 10 years ago. The remaining 4 patients were aged between 10 and 19 years old, accompanied by varying degrees of ametropia and amblyopia. They were currently undergoing a one-stage surgical treatment (epicanthus correction and eyelid fissure enlargement). This finding also confirmed that patients with BPES can have amblyopia and ametropia at the same time. However, due to the small number of cases, a detailed description of the visual function of patients with BPES could not be made. Since BPES has a serious impact on the visual function of children, early diagnosis and treatment of congenital ptosis will help to prevent and manage these eye abnormalities.
In conclusion, we identified a novel FOXL2 insertion mutation c.672_701ins GCGGCTGCCGCCGCAGCTGCTGCA GGCGCT (p.Ala234_Gly235linsAAAAAAAAGA) in a Chinese family with BPES type II. The mutation was located at the polyalanine tract and first reported in the Asian people.The phenotypes of the pedigree included small palpebral fissures, ptosis, telecanthus and epicanthus inversus with normal fertility. The present study amplified the genotypic spectrum of FOXL2-BPES and better illustrates its genotypephenotype correlations, which provided a basis for elucidating the pathogenesis of BPES and genetic counseling.
ACKNOWLEDGEMENTS
Conflicts of Interest: Rong WN,None;Ma MJ,None;Yang W,None;Yuan SQ,None;Sheng XL,None.
 International Journal of Ophthalmology2021年4期
International Journal of Ophthalmology2021年4期
- International Journal of Ophthalmology的其它文章
- Prevalence and risk factors of dry eye disease in young and middle-aged office employee: a Xi’an Study
- YM155 inhibits retinal pigment epithelium cell survival through EGFR/MAPK signaling pathway
- Clinical features and treatment outcomes of intraocular lymphoma: a single-center experience in China
- Trends in research related to high myopia from 2010 to 2019: a bibliometric and knowledge mapping analysis
- A simple new technique for the induction of residual posterior vitreous cortex removal and membrane peeling
- Differential degeneration of rod/cone bipolar cells during retinal degeneration in Royal College of Surgeons rats