
共表达网络鉴定食管鳞癌与腺癌关键差异表达分子
2021-04-07 00:32:08李孟祥刘轲阮豪杰高社干齐义军冯笑山
暨南大学学报(自然科学与医学版) 2021年2期
李孟祥, 刘轲, 阮豪杰, 高社干, 齐义军*, 冯笑山
(1.河南科技大学 信息工程学院,河南 洛阳 471023;2.河南科技大学 临床医学院/第一附属医院/河南省肿瘤表观遗传重点实验室,河南 洛阳 471003)
食管癌(esophageal cancer, EC)位居中国恶性肿瘤发病率和死亡率的第4位,中国每年确诊的EC病例和死亡病例约占全球总确诊病例和死亡病例的一半[1].2015年,中国EC的新发病例、新增死亡病例约为24.6万例、18.8万例[2].EC主要有两种组织病理学类型:食管鳞状细胞癌(esophageal squamous cell carcinoma, ESCC)和食管腺癌(esophageal adenocarcinoma, EAC).在中国,至少90%的EC为ESCC,而欧美等西方国家却以EAC为主[3-4].
ESCC大多发生在食管中、上段,病因学上与抽烟、酗酒等密切相关.EAC主要发生于食管下段与贲门交界处,病因学上与肥胖、胃食管反流等关系密切.此外,ESCC与肺、宫颈等组织部位的鳞癌基因突变模式类似[5],而EAC和胃腺癌分子病变模式相近.因此,鉴定起源于食管但病理类型不同的ESCC与EAC特异性分子表达谱,是靶向治疗和个体化治疗的基础.
加权基因共表达网络分析(weighted gene co-expression network analysis, WGCNA)通过构建共表达网络确定基因模块,分析模块与性状表型之间的相关性,从而挖掘与临床表型相关的关键分子[6].本研究应用公共数据库中EC的mRNA表达谱测序数据,鉴定EAC和ESCC差异表达基因,然后应用WGCNA算法构建基因共表达网络模块,确定模块内关键基因以鉴别EAC和ESCC并进行验证.
1 材料和方法
1.1 EC的mRNA表达谱数据
从癌症基因图谱数据库(the cancer genome atlas, TCGA)和基因表达数据库(gene expression omnibus, GEO)下载EC的mRNA转录组数据及相应临床信息,分别为TCGA-EC和GSE26866.将表达谱数据与临床信息匹配后,共纳入来源于TCGA-EC的78例EAC和80例ESCC;GSE26866数据集包括20例Barrett’s食管(Barrett’s esophagus, BE)、21例EAC、9例ESCC和19例食管鳞状上皮(esophageal squamous epithelium, ESE)样本(表1).

表1 入组数据集的组织类型和样本数量
1.2 TCGA-EC差异表达基因鉴定
用R语言中DESeq2程序包处理TCGA-EC mRNA表达谱数据,差异表达基因筛选条件为:差异倍数(fold change, FC)>2,错误发现率(false discovery rate, FDR)<0.05且base mean >10;然后将所有基因表达值进行归一化处理,确定适合样本相关性计算的基因表达谱数据.
1.3 WGCNA网络模块构建
在WGCNA构建的基因共表达网络中,同一个模块内的基因表达模式相似,而不同模块的基因表达模式差别较大,通过加权使得整个网络的连接服从无标度网络分布[7].本研究在构建无标度网络时,确定的软阈值β须满足:R2>0.9,且平均连接度小于100.通过名为“WGCNA”的程序包构建基因聚类树,并采用动态剪切算法鉴定基因模块[8].
根据模块第一主成分Module Eigengenes(ME)计算模块内所有基因与ME的相关性,确定基因的模块隶属度(module membership, MM);根据ME与EAC、ESCC表型的相关性,确定基因显著性(gene significance, GS);MM与GS均满足一定条件的基因为模块的节点基因.
1.4 富集分析及独立验证
对重点模块的节点基因进行GO和KEGG富集分析,其中GO富集分析包括细胞成分(cellular component, CC)、生物学过程(biological process, BP)和分子功能(molecular function, MF);KEGG富集分析则是在所有已知的生物学通路(pathway)中进行.根据超几何分布检验显著性FDR,确定模块内显著富集的生物学功能和通路.使用ClusterProfiler程序包进行富集分析及相关绘图[9].
应用3组独立数据验证上述关键基因鉴别ESCC和EAC的准确性,分别是GSE26866、肺鳞状细胞癌(lung squamous cell carcinoma, LUSC)和肺腺癌(lung adenocarcinoma, LUAD)、癌细胞数据库(cancer cell line encyclopedia, CCLE)中23个ESCC细胞系和1个EAC细胞系.
1.5 统计学分析
用R语言(3.6.3版本)进行统计学分析.两组样本表达谱差异比较,采用Wilcoxon检验;多组样本表达谱差异比较,采用Kruskal-Wallis检验,并进行P值的FDR校正.以P<0.05或FDR<0.05为差异有统计学意义.
2 结果
2.1 EAC和ESCC差异表达基因
根据本研究确定的差异表达基因筛选标准,分析TCGA数据集中EAC和ESCC基因表达数据,共鉴定到EAC和ESCC的差异表达基因3745个,ESCC中上调和下调的基因个数分别为1 885个(50.33%)和1 860个(49.67%,图1).
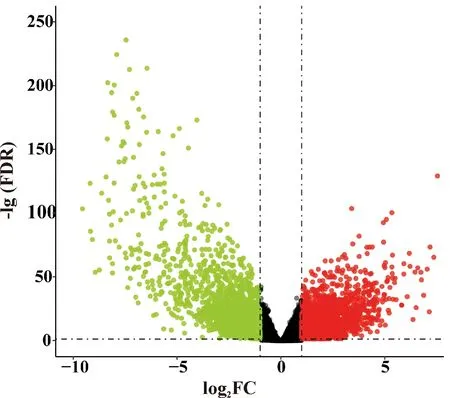
红色和绿色分别代表食管鳞癌中上调和下调的基因Red and green represent up-regulated and down-regulated genes in esophageal squamous cell carcinoma, respectively图1 TCGA数据集中食管鳞癌与食管腺癌差异表达基因火山图Fig.1 Volcano map of DEGs in esophageal squamous cell carcinoma and esophageal adenocarcinoma in the TCGA dataset
2.2 共表达网络模块分析
基于ESCC和EAC的3745个差异表达基因,对软阈值β取1~20的网络拓扑进行分析,在标度独立性和平均连通性条件下选择β=5,此时网络无标度拓扑拟合指数大于0.9且平均连接度小于100,网络结构近似于无标度网络.通过动态剪切树算法将相似性大于0.8的模块合并为同一个模块,最终共得到18个模块,其余不能归类到上述18个模块的304个基因归为灰色(grey)模块(图2).

上方为基因聚类图谱;下方为动态剪切后基因所属模块,不同颜色代表不同模块.The upper part is the gene clustering map and the bottom is the module to which the gene belongs after dynamic shearing with different colors denoting different modules图2 食管鳞癌和食管腺癌差异表达基因和共表达网络模块分布Fig.2 Distribution of differentially expressed genes between esophageal squamous cell carcinoma and esophageal adenocarcinoma, and co-expression network modules by WGCNA
应用相关性分析计算19个模块中每个模块ME与ESCC/EAC、BMI、性别、年龄和临床分期的相关性,图3显示19个模块在4种不同表型中的分布.与其他3个表型特征相比,不同组织学类型食管肿瘤(ESCC vs EAC)与模块的相关性最强,其中9个模块与ESCC呈正相关,10个模块与EAC呈正相关.19个模块与BMI的相关性仅次于组织学类型,令人感兴趣的是,19个模块在食管肿瘤的组织学类型与BMI中的分布完全相反,与ESCC呈正相关的模块则与BMI呈负相关,反之亦然.19个模块在年龄特征和临床分期特征中分布与BMI相似,但与ESCC/EAC呈负相关,但相关性低于BMI.所有模块与性别相关性系数均介于-0.2到0.11之间,相关性较小(图3).
与EAC呈正相关的10个模块中,Blue模块的相关性最大(cor =-0.95,P=7e-79),该模块共包含1 265个基因,这些基因在EAC中高表达;而Turquoise模块与ESCC呈正相关(cor=0.89,P=9e-55),该模块中的1 454个基因在ESCC中高表达.4a和4b分别显示了Blue和Turquoise模块内所有基因的模块隶属度MM与基因显著性GS的分布关系.
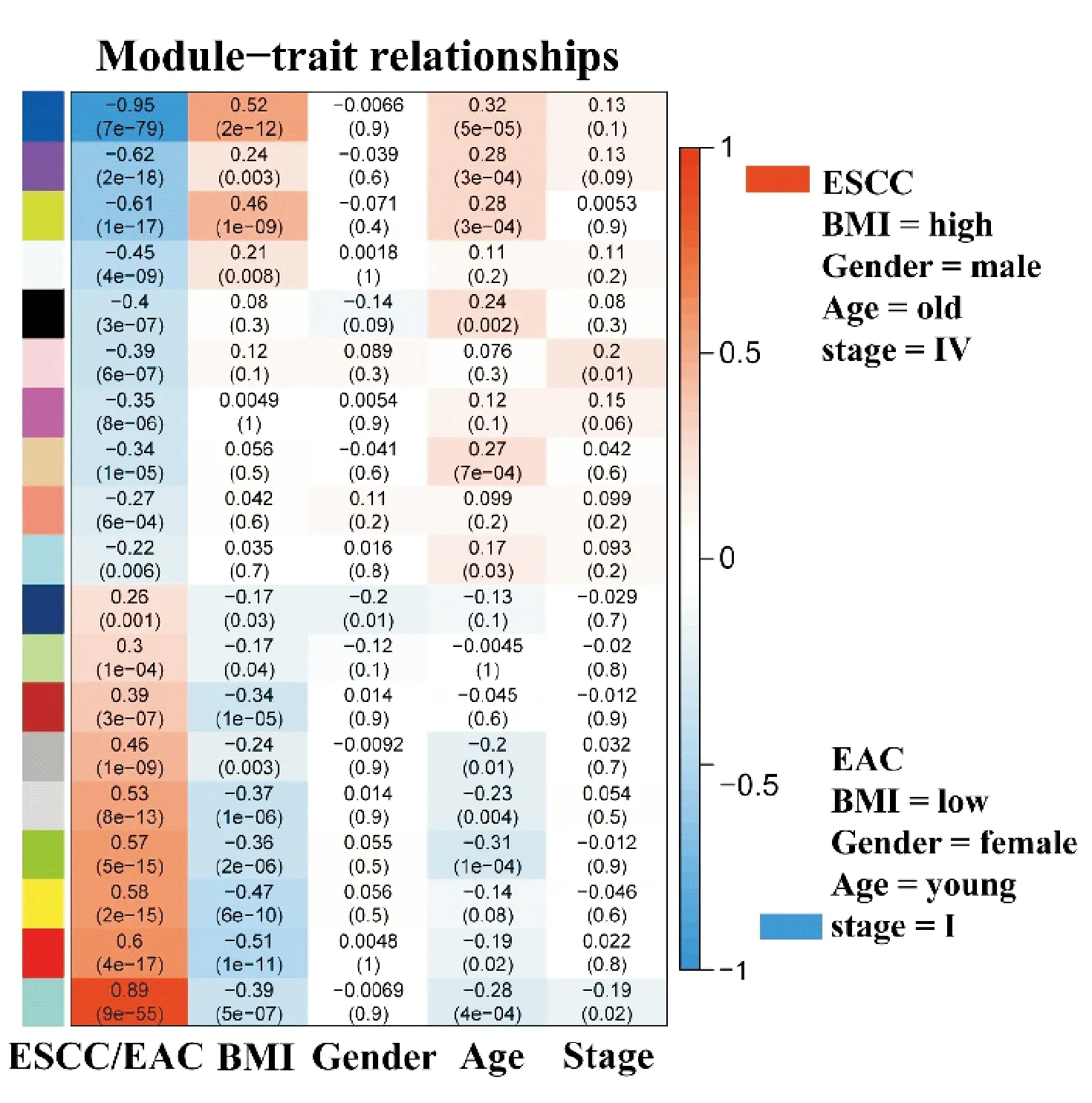
图3 模块与ESCC/EAC、BMI、Gender、Age、 Stage(肿瘤临床分期)的相关性热图Fig.3 Correlations between heat map of modules and ESCC/EAC, BMI, Gender, Age, Stage
2.3 EAC和ESCC代表性基因确定及验证
分别计算Blue模块和Turquoise模块中构成基因的MM与GS之和,以确定每个模块中的节点基因.结果表明,EPS8L3和SOX15分别是Blue模块和Turquoise模块中MM与GS之和最大的基因,即最重要的节点基因,因此确定EPS8L3和SOX15分别为EAC和ESCC的代表性基因.
为验证本实验确定的EAC和ESCC代表性基因,本实验应用3组不同来源的样本进行验证.在TCGA的80例ESCC和78例EAC样本中,EPS8L3在EAC和ESCC中的表达值分别为12.83±1.21、5.87±0.86(P<0.000 1,图4c),SOX15的表达值分别为7.91±1.47、12.34±1.16(P<0.000 1,图4d).在GSE26866数据集中,EPS8L3在21例EAC中的表达明显高于ESCC(校正后P<0.001,图4e),而9例ESCC组织中SOX15的表达明显高于EAC(校正后P<0.001,图4f),EPS8L3和SOX15在ESCC中的表达值分别是-3.98±0.82、3.15±1.99.同样,EPS8L3在EAC细胞系OE19中表达值为48.89,显著高于23株ESCC细胞系中的平均表达值0.059;相反,SOX15在23株ESCC细胞系中平均表达值明显高于OE19细胞系(25.72和0.335).为进一步验证EPS8L3和SOX15是腺上皮和鳞状上皮的特异性标志物分子,本实验应用TCGA数据库中的498例LUSC和476例LUAD做进一步验证,EPS8L3和SOX15在LUSC和LUAD中的表达模式与ESCC和EAC一致(P<0.01,图4g;P<0.01,图4h).上述实验结果表明,EPS8L3和SOX15分别是上皮来源的腺癌和鳞状细胞癌特异性分子,可用于二者的鉴别诊断.
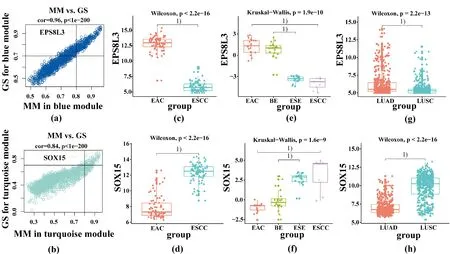
(a-b): blue和turquoise模块内基因的MM与GS特征散点图; (c-d): EPS8L3和SOX15在数据集TCGA中的表达值比较; (e-f): EPS8L3和SOX15在数据集GSE26886中的表达谱比较,差异显著性为FDR法矫正后的P值; (g-h): EPS8L3和SOX15在数据集LUSC、LUAD中的表达谱比较. 两组样本比较用Wilcoxon检验;多组样本比较用Kruskal-Wallis检验;1)P<0.001.(a-b): Scatter plots of MM and GS characteristics of genes in blue and turquoise module; (c-d): Comparison of expression profiles of EPS8L3 and SOX15 in the data sets ESCC and EAC; (e-f): Comparison of expression profiles of EPS8L3 and SOX15 in data set GSE26886, the P value was corrected by the FDR method; (g-h): Comparison of expression profiles of EPS8L3 and SOX15 in data sets LUSC and LUAD. The Wilcoxon test was used for the comparison of two groups of samples; the Kruskal-Wallis test was used for the comparison of multiple groups of samples; 1) P<0.001.图4 关键节点基因表达值比较Fig.4 Comparation of key hub genes from modules and their expression values.
2.4 基因生物学功能和通路富集分析
以MM>0.8和GS>0.7为标准筛选模块节点基因,Blue模块和Turquoise模块分别鉴定了259个和66个节点基因.对Blue模块中259个基因做GO富集分析,结果表明这些基因的功能主要富集于细胞级性(Apical part of cell)、肌动蛋白相关的细胞形态(Actin-based cell projection)等(图5a).对Turquoise模块66个节点基因的GO和KEGG富集分析显示,这些基因主要富集于表皮发育(epidermis development)、角质细胞分化(keratinocyte differentiation)、表皮细胞分化(epidermal cell differentiation)、雌激素信号通路(estrogen signaling pathway)、Rap1信号通路(rap1 signaling pathway)等生物学功能(图5b).

(a): Blue模块GO富集分析结果; (b): Turquoise模块的GO富集分析结果.(a): GO enrichment analysis result of blue module; (b): GO enrichment analysis result of turquoise module.图5 Blue和Turquoise模块组成基因富集分析Fig.5 Enrichment analysis of blue and turquoise
3 讨论
在病理类型、病因学、好发部位等方面,中国与欧美等西方国家EC完全不同[10],因此,我国以ESCC为主的临床治疗方案不能完全借鉴欧美等国家EAC为主的治疗方案,亟需制定适合中国EC,尤其是ESCC的治疗原则及有效治疗方案.
应用芯片及高能量测序技术和生物信息学方法鉴定不同肿瘤、同一部位不同组织类型的肿瘤,能够为肿瘤的精准治疗提供有效的分子靶点.有研究表明[11],SOX2、PIK3CA、MYC、CCND1、FGR1、GATA4、GATA6等基因位点扩增在ESCC和EAC中明显不同,含有SOX2和PIK3CA基因位点的3q在ESCC中扩增频率显著高于EAC中(60% vs 15%).应用外显子测序数据分析在美国ESCC和EAC中的热点基因的突变频率,发现NOTCH1失活突变在ESCC中存在较高的突变频率(21%),而EAC中不存在NOTCH1的失活突变.进一步比较美国和中国ESCC热点基因分子差异,发现美国ESCC中NOTCH1的失活突变频率远远高于中国的ESCC(21% vs 2%),但TP53却没有明显差异[12].Wang等[13]通过二代测序数据分析比较了ESCC和EAC样本在突变、插入缺失、基因扩增、纯合缺失等基因组改变的异同,发现在临床相关基因组改变中,EAC样本中KRAS、ERBB2改变更为频繁,而ESCC样本中PIK3CA、PTEN、NOTCH1等改变频率更高.尽管如此,ESCC和EAC中也存在相同的分子变化,如CDKN2A、EGFR、KRAS、MYC、CDK6和MET等基因位点扩增同等的发生于ESCC和EAC.
基于表达模式相近的基因具有相似的生物学功能或相同的调节机制的观点[14],WGCNA利用分子间加权表达相关性,衡量不同分子的表达模式关系.由此,一方面可以近似还原分子相互作用的无标度网络分布关系,另一方面将复杂的分子表达数据简化为若干功能模块,以挖掘表达模式相似及具有相同生物学功能的分子.WGCNA通过构建分子共表达模块,常用于鉴定与疾病进展、复发、预后相关的分子功能模块[15].应用WGCNA分析TCGA数据库中平滑肌瘤测序数据[16],构建24个功能模块,发现其中的Dark red模块与疾病复发呈正相关(cor=0.32,P=9e-4),进一步用Cytoscape软件插件CytoHubba鉴定该模块的12个节点基因,结合平滑肌瘤患者的临床生存数据,最终确定12个基因中的3个基因(CDK4、CCT2和MGAT1)是平滑肌瘤的特异性表达基因,并应用ONCOMINE数据库中的平滑肌瘤数据进行了外部验证.同样,利用WGCNA对16 383个肝细胞癌差异表达基因构建25个模块[17],并分析这25个模块与肝细胞癌(HCC)的癌变过程(正常肝、无HCC的肝硬化、肝硬化、低度发育异常、高级别发育异常、极早期和早期、晚期HCC和极晚期HCC)的相关性,发现5个节点基因(GINS1、TOP2A、BUB1B、ACADM和ARPC4)是肝细胞癌的预后分子标志物.
本研究基于EC的mRNA表达谱数据,应用WGCNA构建了19个模块,其中的Blue和Turquoise模块分别与EAC、ESCC具有显著的相关性.Blue模块中的基因在EAC中表达较高,而Turquoise模块中的基因在ESCC中明显高表达.此外还发现Blue模块与BMI呈正相关(cor=0.52,P<0.001),这与之前研究结果相一致[18].由于BMI较高的肥胖人群导致胃食管反流,使罹患EAC风险增加,表明WGCNA鉴定的Blue模块及组成基因是EAC和ESCC具有不同发病机制的分子基础.
通过MM与GS之和确定节点基因的重要性,本研究发现EPS8L3和SOX15分别是EAC和ESCC的代表性基因,并应用3个不同的数据集验证其准确性.不仅如此,EPS8L3和SOX15还能区分LUSC和LUAD,进一步表明这两个分子具有不同的组织起源,是鉴别鳞状上皮或腺上皮来源肿瘤的特异性分子,为鳞癌和腺癌的临床治疗方案选择提供理论基础.EPS8L3与多种肿瘤发生发展、化疗敏感性相关,如肝癌组织和细胞系中EPS8L3高表达并与肝癌患者的预后不良有显著相关性[19](P<0.011),EPL8L3基因沉默能够增加对顺铂的敏感性.SOX15是一种必要转录因子,高表达于LUSC、睾丸生殖细胞肿瘤、胸腺瘤等,主要通过调控WNT/β-catenin信号通路参与多种肿瘤的发生、发展[20].SOX15基因沉默能够降低WNT/β-catenin信号通路活性,抑制ESCC细胞增殖、增加ESCC对紫杉醇的敏感性[21].上述结果表明,EPS8L3和SOX15不仅能够甄别ESCC和EAC,还是潜在分子治疗靶点.
本研究应用WGCNA构建ESCC和EAC差异表达基因的共表达网络,鉴定ESCC和EAC特异表达基因SOX15和EPS8L3,能够鉴别起源于食管或肺组织的肿瘤组织类型,并有助于ESCC和EAC的临床治疗方案制定.
猜你喜欢
中老年保健(2021年9期)2021-08-24 03:49:56
昆明医科大学学报(2020年12期)2021-01-26 00:44:02
农村青少年科学探究(2020年5期)2020-08-18 02:20:52
新民周刊(2018年8期)2018-03-02 15:45:54
饮食科学(2017年12期)2018-01-02 09:23:20
山东医药(2017年20期)2017-07-01 19:52:07
中国学术期刊文摘(2016年1期)2016-02-13 14:05:23
健康管理(2015年3期)2015-11-20 18:22:36
少儿科学周刊·少年版(2015年1期)2015-07-07 21:57:30
山东医药(2015年44期)2015-02-28 14:29:02
