
La-C共掺ZnO 的电子结构和光学性质的第一性原理研究
2021-03-22 09:58赵志峰王海芳张天玑
电子元件与材料 2021年2期
赵志峰,王海芳,张天玑,肖 磊
(中北大学 环境与安全工程学院,山西 太原 030051)
氧化锌(ZnO)是一种新型的Ⅱ-Ⅵ族直隙氧化物半导体材料,禁带宽度为3.37 eV,激子结合能为60 meV[1-2]。因其优越的化学稳定性和优良的光电性能,其不仅在光电器件中具有良好的应用前景[3-5],同时也在光催化领域获得越来越多的关注[6-7]。但由于ZnO的禁带宽度较大,只能吸收占太阳光3%~5%,对太阳光的可利用范围小,这限制了ZnO的光催化活性[8-9]。
研究表明,通过掺杂可以调节ZnO 的禁带宽度,对能带结构进行修饰或者形成杂质能级,提高电子跃迁的效率和概率,同时抑制光生载流子的复合,提高ZnO的光催化活性。为此人们进行了许多尝试,非金属掺杂方面,研究人员研究了N、C、S等非金属元素掺杂ZnO的光电性质,发现掺杂后ZnO的禁带宽度会变小,电子空穴对的复合速率变小,光催化活性提高[9-13]。金属掺杂方面,稀土元素由于其独特的电子结构和光谱性质,目前已成为研究ZnO掺杂体系的热点,研究发现La、Nd、Pr等稀土元素掺杂ZnO会形成晶格缺陷,影响光生电子和空穴的运动状况,改变ZnO的能带结构,但是金属掺杂后晶体的稳定性差[14-15]。结合金属和非金属掺杂的优缺点,Yamamoto等通过第一性原理方法对电子结构进行研究,表明通过金属元素和非金属元素共掺,能增加受主的掺杂浓度,获得较浅的受主能级[16],如La-N、B-P、Cu-C共掺杂ZnO等[16-19],阴阳离子共掺杂ZnO离子间的协同作用进一步提高ZnO的光吸收范围和吸收活性。Youssef等采用溶胶凝胶法制备了La-C共掺杂ZnO纳米颗粒,发现ZnO纳米颗粒增强了对亚甲基蓝降解的光催化活性[20],但目前对于其微观机理的研究很少。
为此本文尝试运用第一性原理计算La-C共掺杂ZnO的能带结构、态密度和光学性质,通过微观机理进行细致分析,期望为La-C共掺杂ZnO 的实验研究工作提供理论参考。
1 计算方法及理论模型
1.1 计算方法
所有计算使用Material Studio 8.0 软件中的CASTEP[21]模块自旋密度泛函理论,计算中由于GGA方法计算体系能带会导致体系的带隙比实验值偏小许多,影响后面的分析,本文将采用GGA+U 的方法进行带隙修正,其中Zn的3d电子U值设置为2.66 eV,O的2p电子U值设置为9.34 eV,La的U 值设置为6.0 eV。计算中对所有超晶胞进行结构优化,优化时选用广义梯度近似GGA 下的PW91交换函数描述电子间相互作用的交互关联能,结构优化时平均每个原子的自洽收敛精度为2.0×10-5eV,平面波截断能为Ecut=420 eV,内应力为0.05 GPa,布里渊区K点选取4×4×2。用于构建赝势的价电子组态分别为Zn3d104s2、O2s22p4、La5s25p65d16s2和C2s22p2。
1.2 理论模型
ZnO晶体结构有4种,分别是闪锌矿结构、纤锌矿结构、八面体结构和正四面体结构。这里使用热力学最稳定的六方纤锌矿型结构,其空间群为P63mc,晶格常数a=b=0.325 nm,c=0.520 nm,角度α=β=90°,γ=120°,基于2×2×2 ZnO超晶胞结构,构建La掺杂ZnO,C掺杂ZnO以及La/C共掺ZnO体系的模型,计算并分析ZnO掺杂体系的能带结构、态密度和光学性质。ZnO掺杂前后的晶胞模型如图1,图1(a)为纯ZnO晶胞模型,图1(b)为La单掺ZnO晶胞模型,使用1个La原子代替1个Zn原子,图1(c)为C单掺ZnO晶胞模型,使用1个C原子替代1个O原子,图1(c)为La-C共掺ZnO晶胞模型,使用1个La原子代替1个Zn原子,1个C原子替代1个O原子。
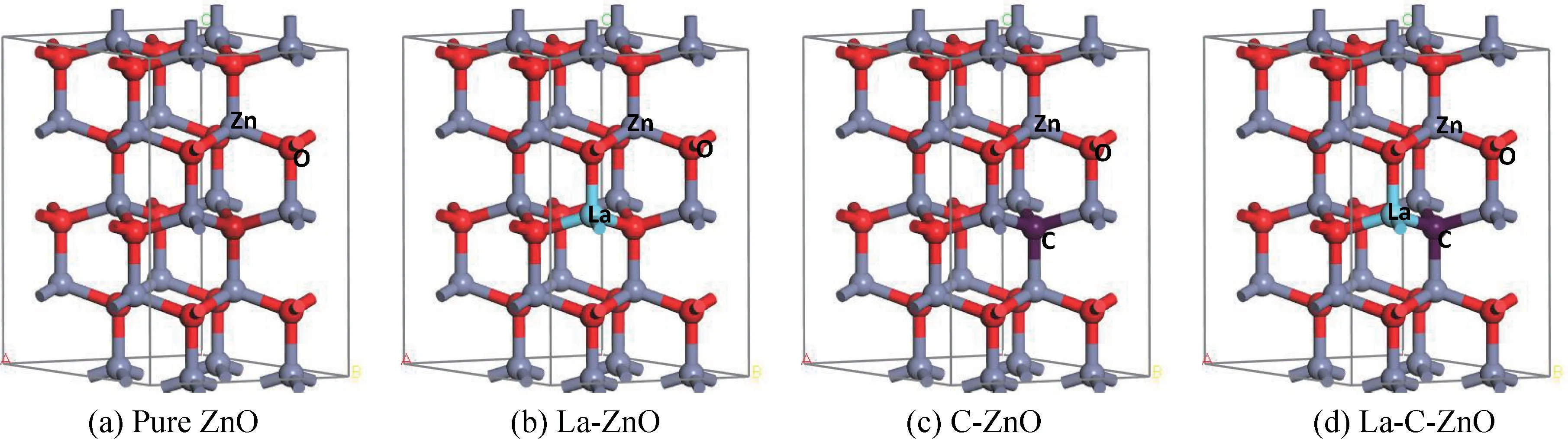
图1 ZnO掺杂前后的晶胞模型Fig.1 Crystal cell model before and after ZnO doping
2 计算结果和讨论
2.1 晶体结构和稳定性分析
对所有掺杂体系进行晶格优化,所得晶格参数如表1所示,可得纯ZnO的晶格常数(a=0.3227 nm,c=0.5118 nm)与实验值(a=0.3253 nm,c=0.5201 nm)[22]相差不大,同时c/a 相差也不大,说明计算参数设置合理,计算结果可信。所有掺杂体系的晶胞体积相对纯晶胞均变大,这是由于掺杂中C原子代替O原子,La原子代替Zn原子,且C原子和La原子的半径分别大于O 原子和Zn原子的半径。这其中C掺杂ZnO的体积和基态能量都最小,说明该体系最稳定。形成能是表征掺杂体系稳定性和原子掺入体系难易程度的物理变量。基于几何结构优化后的体系总能量和不同原子的化学势计算相应结构的形成能。各掺杂体系的形成能Ef满足公式(1)-(3)。
La掺杂ZnO体系形成能:
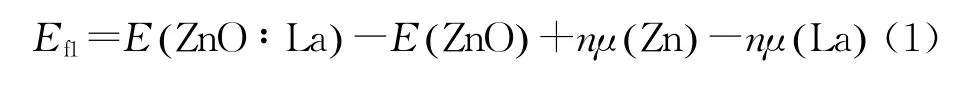
C掺杂ZnO体系形成能:

La-C共掺杂ZnO的形成能:
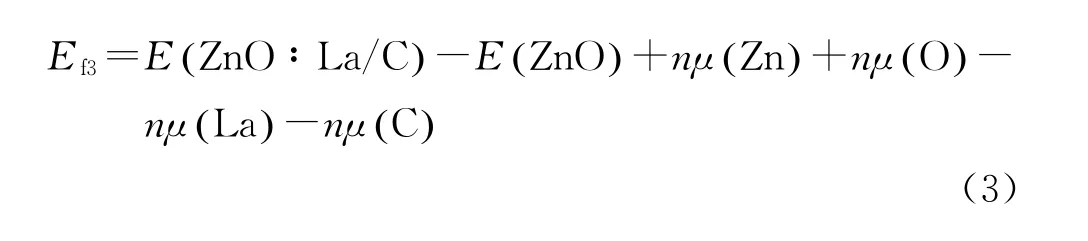
式中:E(ZnO∶La)、E(ZnO∶C)、E(ZnO∶La/C)表示各掺杂体系掺杂后的体系总能量;E(ZnO)表示与掺杂体系相同尺寸的纯ZnO超晶胞体系总能量;系数n 表示掺入的原子和替代的原子数;μ(Zn)、μ(La)、μ(O)、μ(C)分别表示各原子的化学势。形成能计算结果表明共掺杂的形成能比单掺杂小,说明共掺杂体系比单掺杂体系更容易形成。

表1 掺杂前后的晶体结构表Tab.1 Crystal structure before and after ZnO doping
2.2 能带结构
图2(a)-(d)分别为ZnO掺杂前后的能带结构图,图中虚线为费米能级,费米能级始终定位0点(Ef=0 eV)。图2(a)为纯ZnO 的能带结构图,计算得到纯ZnO的禁带宽度为3.368 eV,这与ZnO 实验值3.37 eV相差不大,说明计算设置的参数是合理的。通过比较可以看出,由于掺杂后晶体发生畸变,对称性下降,同时导带数和价带数增多,能带结构变得更加紧凑,区域内的电子曲线变得密集,这说明掺杂后区域间的电子间相互作用变强。图2(b)为La掺杂ZnO的能带结构图,与图2(a)相比,导带和价带均向下移动,禁带宽度变小,带隙值变为3.12 eV,缩小了电子从价带顶跃迁到导带底所需要的能量。图2(c),图2(d)分别为C掺杂ZnO的能带结构图和La-C共掺ZnO的能带结构图,发现图中都出现杂质能级,这两个掺杂体系价带顶和导带底的间距分别为3.658 eV 和3.708 eV,掺杂体系杂质能级与价带顶和导带底之间的禁带宽度分别为0.634 eV,2.182 eV 和1.03 eV,1.713 eV。杂质能级的出现为电子从价带跃迁到导带建立了桥梁,同时由于能带带隙变小,使得电子容易激发从价带跃迁到杂质层,再跃迁到导带,使得电子从价带跃迁到导带的过程变得容易,且杂质能级的存在可能会阻碍电子和空穴的复合,有利于提高ZnO的光催化性能。
2.3 态密度
图3(a)-3(d)分别为ZnO掺杂前后的总态密度图和分波态密度图,取费米能级为能量零点,发现掺杂体系与纯ZnO 比较,总态密度整体向低能方向移动。图3(a)为纯ZnO的态密度图,可以看出未掺杂的ZnO的导带主要由Zn的3p态和O的2p态贡献,价带主要由Zn的3d态和O的2p态贡献,其中O的2p态贡献最大。图3(b)为La单掺ZnO的态密度图,导带主要由Zn的3d态,O的2p态以及La的5d态贡献,价带则主要是由Zn的3d态和O的2p态贡献,费米能级出现杂质能级,主要由La的5d态贡献。图3(c)为C单掺ZnO的态密度图,费米能级附近出现新的态密度峰,这出现的杂质能级主要由C的2p态贡献,同时发现价带和导带的主要贡献能级基本未发生变化,表明C的掺杂对能带有明显的影响,其导致费米能级附近出现杂质能级。图3(d)为La-C共掺ZnO的态密度图,费米能级附近出现的杂质能级主要是La的5d态和C的2p态贡献,C的2p态贡献大于La的5d态。掺杂体系相比较于纯ZnO,由于禁带宽度变小和杂质能级的产生,导电性增强,光催化性能提升。

图2 能带结构图。(a)Pure ZnO;(b)La-ZnO;(c)C-ZnO;(d)La-C-ZnOFig.2 Band structure diagram.(a)Pure ZnO;(b)La-ZnO;(c)C-ZnO;(d)La-C-ZnO

图3 能态密度图。(a)Pure ZnO;(b)La-ZnO;(c)C-ZnO;(d)La-C-ZnOFig.3 Energy state density diagram.(a)Pure ZnO;(b)La-ZnO;(c)C-ZnO;(d)La-C-ZnO
2.4 光学性质
图4(a)和图4(b)分别是ZnO 掺杂前后的吸收光谱图和局部吸收光谱图,得到纯ZnO 体系、La掺杂ZnO体系、C掺杂ZnO 体系、La-C共掺杂ZnO 体系的第一个吸收峰分别位于4.38,2.34,1.09和2.27 eV处。通过掺杂体系的第一个吸收峰位置发现掺杂使得ZnO的光吸收范围扩展到了可见光区域1.64~3.19 eV,同时与纯ZnO相比,可以看出所有掺杂体系的吸收带边都发生了明显红移,有利于提高ZnO的光催化能力。分析认为吸收带边红移的原因是由于掺杂后禁带宽度变小,减少了电子从价带到导带跃迁所需要的能量,从而诱发吸收光谱发生红移。
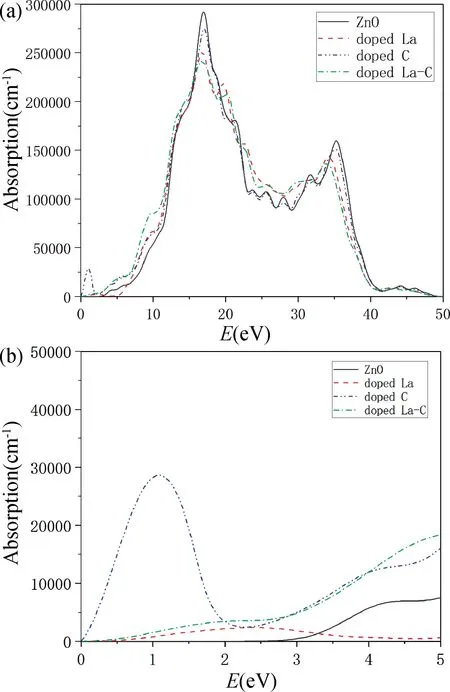
图4 光学性质图。(a)吸收光谱;(b)低能区吸收光谱Fig.4 Optical properties.(a)Adsorption spectra;(b)Adsorption spectra in low region
图5(a)为ZnO 掺杂前后的介电函数的实部曲线图,在无入射光的情况下,介电函数实部对应的纵坐标为静态介电常数。从图5(a)中可以得到纯ZnO,La单掺ZnO,C单掺ZnO,La-C共掺ZnO的静态介电常数分别为2.338,2.5783,19.297,3.265。和纯ZnO相比,发现所有掺杂体系的静态介电常数变大,说明掺杂后ZnO 的极化能力变强,对电荷的束缚能力变强,掺杂体系的光生电场强度变大,光激发载流子在晶体内的迁移变快。图5(b)为ZnO掺杂前后的介电函数的虚部曲线图,表征的是形成电偶极子消耗的能量,反映了能带结构和光跃迁之间的关系。从图中可以看出纯ZnO 的三个主峰位于4.324,13.108和16.209 eV 处,La单掺ZnO 体系的三个主峰位于1.664,13.142和16.086 eV处,C单掺ZnO体系的三个主峰位于0.321,13.188和16.139 eV 处,La-C共掺ZnO体系的四个主峰位于1.5484,4.789,9.273和12.841 eV处。和纯ZnO 相比,发现掺杂体系的主峰均向低能区移动,这与掺杂后晶胞发生畸变,掺杂体系的最小禁带宽度均变小相一致。
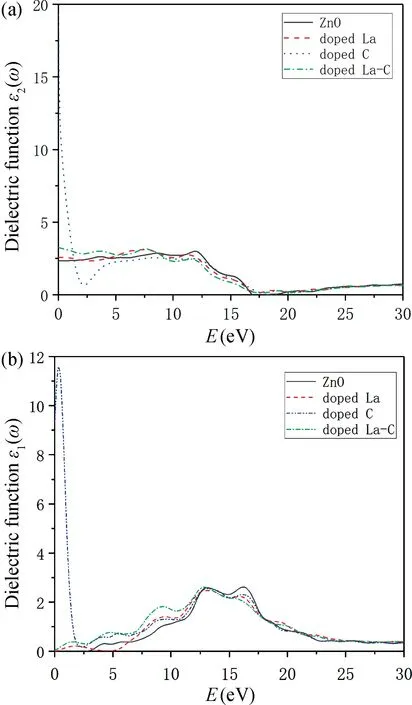
图5 光学性质图。(a)介电函数实部;(b)介电函数虚部Fig.5 Optical properties.(a)The real part of the dielectric function;(b)The imaginary part of the dielectric function
3 结论
本文基于密度泛函理论的第一性原理,以ZnO为本征体,建立2×2×2超晶胞,研究了La单掺ZnO,C单掺ZnO以及La-C共掺ZnO 的电子结构和光学性质,得出以下结论:
(1)所有掺杂体系结构优化后,晶胞体积均变大,形成能小于0,且La-C共掺ZnO的形成能最小,说明共掺杂体系比单掺杂体系更容易形成。
(2)在电子结构方面,与本征ZnO 相比,La单掺ZnO的禁带宽度变小,C单掺ZnO 和La-C共掺ZnO的能带结构均出现了杂质能级,杂质能级的出现为电子的跃迁提供了桥梁,有利于提高ZnO 的光催化性能。
(3)由光学性质分析可知,与本征ZnO 相比,所有掺杂体系的吸收带均发生了红移,对可见光的响应范围变大,提升了ZnO的光催化性能。掺杂后所有体系的静电常数变大,说明掺杂后极化能力变强,体系对电荷的束缚能力变强。
猜你喜欢
高中数理化(2022年16期)2022-09-14
高中数理化(2022年4期)2022-03-14
装备维修技术(2021年36期)2021-10-25
弹箭与制导学报(2021年3期)2021-07-30
中学生数理化(高中版.高考理化)(2019年10期)2019-11-08
网印工业(2019年4期)2019-05-21
重型机械(2019年2期)2019-04-28
山东化工(2019年1期)2019-01-24
吉首大学学报(自然科学版)(2018年3期)2018-07-03
