
群体遗传学下动物驯化研究进展
2021-03-20 01:02文子龙赵毅强
遗传 2021年3期
文子龙,赵毅强
综 述
群体遗传学下动物驯化研究进展
文子龙,赵毅强
中国农业大学生物学院,北京 100193
动物驯化是将野生动物改变为能够长期稳定饲养的家养动物的过程。作为新石器时代农业革命的内容,驯化是人类社会文明进步的重要标志之一。由于和人类的密切关系,驯化不仅改变了动物的野生状态,也改变了人类的生活习性和文明进程。动物驯化研究的关键问题包含驯化祖先是谁、驯化所产生的改变及驯化时间地点等。随着高通量基因组技术和对应分析方法的发展,目前研究动物驯化一般基于群体水平,在群体遗传学的框架下研究动物驯化过程中的重要事件。本文总结了群体遗传学下动物驯化研究的相关内容,包括群体动态历史、选择信号、基因交流等,着重介绍了基因选择初始时间和基因交流时间两个新的拓展内容及分析方法,概述了家猪()、家鸡()、绵羊()和山羊()等几种主要农业动物近期驯化研究的进展,以期为动物驯化研究提供了新的方向和视角。
驯化;家养动物;群体遗传学;选择初始时间;基因交流时间
动物驯化是人类根据自身的需求将动物从野生状态改变为豢养状态并为人类提供所需的过程。在驯化过程中,野生动物由凶猛变为温顺,从低产变为高产等[1]。这一过程会持续多个世代[2]。距今约12,000多年前,人类逐渐抛弃了通过采集和狩猎获取食物,而慢慢转向以农耕和驯养动物为主的生产方式[3]。驯化是新石器时代农业革命的重要内容,是人类社会文明进步的标志之一。研究表明驯化事件主要发生在世界上的9个地区:中东、中国、美国东部、中美洲、安第斯/亚马逊、西非热带、萨赫勒、埃塞俄比亚和新几内亚[4]。动物驯化是人类进行的最广泛的遗传实验,驯化导致动物的某些性状,如性情、生长和繁殖等方面与野生祖先相比产生了显著的改变[5,6]。通过开展动物驯化的研究,不仅有助于人们了解野生动物的迁移、演化规律和遗传多样性,还能帮助人们更全面的了解现代人类文明社会的发展历史和传染病历史[7]。
对于动物驯化研究,最基本的问题在于驯化物种的野生祖先有哪些?由驯化引起哪些基因组和表型的变化?以及驯化所发生的时间和地点等[5,6,8]。考古学是研究动物驯化历史的一个重要方法。经过考古发现,对获得的各种动物遗骸进行骨骼形态学、同位素断代法等分析,从而达到追踪其驯化轨迹,了解其驯化起源和驯化历史的目的。考古学相关研究给研究者提供了人类驯养动物的直接证据。例如,在近东和欧洲东南地区的坟墓中发现的陶罐证明人类在7000年之前就已经饮用牛奶[7,9]。此外,在哈萨克斯坦地区的Eneolithic Botai遗址中发现了可以追溯到5500年前的一具马骨架上有系过缰绳的印迹[10]。虽然考古学常用的C14法能够精确的判断时间范围,但其只能对已经发掘出来的驯化动物标本进行推断,加上驯化之初的家养动物与野生祖先在形体状态上差异较小,如狼与早期家犬就具有相似外形,使得其推断的起源时间并不是驯化动物的真实起始时间,且该时间会比真实的驯化起始时间要晚[7]。
群体遗传学兴起于20世纪初,是遗传学的分支领域,也是研究生物进化的主要手段[11]。群体遗传学主要关注种群内部和种群之间的遗传差异研究,其中包括自然选择(natural selection)、人工选择(artificial selection)、漂变(gene drift)、突变(mutation)、基因流(gene flow)及群体历史动态(demography)等过程的研究。群体遗传学现在正处于一个前所未有的革命性阶段,随着DNA测序技术的高速发展,遗传数据被快速、准确、大量地产生。目前动物驯化研究主要在群体基因组水平,使用遗传多态标记来研究家养动物与其野生祖先之间的关系以及可能的驯化事件,其相关的研究方法也就此产生且快速发展[12]。用于动物驯化研究的遗传多态性标记包括微卫星标记、线粒体变异及以SNP为主的基因组变异[13]。2016年,Wang等[14]对总共58只犬科动物的全基因组进行大规模重测序,其中包括12只灰狼()、27只来自亚洲和非洲的土狗()以及分布世界各地的19种不同犬种。分析得出:家犬()起源于3.3万年前的东亚南部,且在15,000年前一部分祖先群体开始迁移到中东、非洲和欧洲,在大约10,000年前到达欧洲。考古学与分子生物学的结合,成为目前研究动物驯化新的方向。Peters等[15]通过对家鸡()基因组数据进行分子钟分析,发现家鸡由野生原鸡()分化而来的时间约在9500±3000年之前,这一时间范围要比考古所得时间早很多。同样,基于全基因组数据所得到的家犬起源时间比考古记录中最早发现的犬化石的时间早了约15,000年[14,16]。
1 群体遗传学下动物驯化研究的经典内容
1.1 群体动态历史估计
群体动态历史是长期和短期变化的综合结果,了解群体动态历史可以帮助我们更好地理解种群在时间维度上的变化,再结合考古或是历史记载则可以估计出精确的动物驯化时间。为了研究物种极其复杂的演化和驯化过程,研究者使用概率论和数理统计设计出了多种基于基因组遗传变异特征的方法重建群体历史[17],包括基于单倍型的方法、基于位点频谱(site frequency spectrum, SFS)、连锁不平衡(linkage disequilibrium, LD)及群体参数模拟等多种方法。
1.1.1 基于单倍型信息
种群的动态事件影响种群的遗传结构,典型的种群动态事件包括种群扩张(expansion)、瓶颈效应(bottleneck)、奠基者效应(founder effect)、群体分化(population subdivision)以及群体间的基因交流等。在溯祖理论的基础上,Li和Durbin[18]首次发表了用于种群动态历史估计的PSMC (pairwise sequentially Markovian coalescent)方法。PSMC是一种隐马尔可夫模型(hidden Markov model, HMM)框架来估计作为隐状态参数的最近共同祖先(time to the most recent common ancestor, TMRCA)的时间,然后使用公式=/4(为群体遗传参数,为突变频率)来计算历史上每个时间点对应的有效群体大小()。该方法认为在二倍体基因组中存在着成千上万个由于重组产生的独立的基因座,每个基因座内的杂合度水平都对应一个TMRCA。有些基因座等位基因间杂合度低,因此共同祖先时间较近,有些基因座等位基因间杂合度高,则其对应的TMRCA较早。根据不同基因座的时间分布,可用来估计种群大小随时间变化的波动。在PSMC基础上,Schiffels等[19]于2014年又扩展了可以同时计算多个个体的多序列马氏链溯祖模型(multiple sequentially Markovian coalescent, MSMC),以及后续开发的版本MSMC2。在估算近期种群动态历史时,MSMC2需要更精准的单倍型分型数据,所以其对单倍型分型错误(phasing error)异常敏感[20]。2016年Terhorst等[20]针对PSMC方法因单个样本且分辨率低、MSMC2方法对单倍型分型错误十分敏感,以及计算负荷重等问题,开发了SMC++ (sequential Markov coalescent + plenty of unlabeled samples)软件。该软件与PSMC都是基于HMM算法与溯祖模型,其还充分利用了SFS及LD信息,SMC++拓展了PSMC使得该方法能够在没有单倍型分型(unphased)的情况下高速处理大量基因组数据,并能够根据位点之间的连锁信息来估算群体之间的分化时间,并推测有效群体大小的变化趋势[20]。为了提高估计的鲁棒性(robustness),PSMC、MSMC2和SMC++均利用自举法(bootstrap)多次进行重复计算来保证估计质量[13]。
1.1.2 基于位点频谱信息
群体变异频率信息也被用于推算群体历史变化。例如,若发现SFS上存在较多低频位点,则可知该群体在某个时间产生了群体扩张事件;而在SFS上低频位点少于预期且整体杂合度偏高时,则推测该群体发生过群体收缩事件。相比于PSMC和MSMC2等方法,基于等位基因变异频率的SFS方法(也叫做mutation frequency spectrum, MFS)[21~23]可以估算更为近期的群体历史,而且可以方便地使用较大的群体信息进行推断。SFS方法需基于先验模型来进行计算,随后未知群体历史参数的后验分布需要从先验分布中采取重复抽样估计所得[24,25]。另外,SFS方法还可以重建十分复杂的多群体历史变化,并推测群体间的迁徙事件。基于SFS方法重塑群体历史的软件包括∂a∂i[24]和Fastsimcoal2[25]。其中,∂a∂i软件使用扩散近似法(diffusion approximation)对一个群体或多个群体的SFS来估算3个群体以内的有效群体大小变化、群体分化及迁移率等;Fastsimcoal2软件则使用复合似然法(composite likelihood)来推算群体的进化过程[13]。
1.1.3 基于连锁不平衡信息
为了克服不完全系谱的限制,越来越多的研究者意识到可以通过基因组数据中的LD信息来估计历史有效群体大小[26]。LD用来描述不同基因座中等位基因的非随机关联关系,其可能由群体历史中的交流事件或基因漂变所产生,或者是源于背景选择(background selection)[27]。因为LD的方差和相关联,所以根据LD可推测[28]。SNeP[26]工具使用SNP数据,并根据LD、和重组率之间的关系估算有效群体大小的趋势,即历史有效群体大小。其公式如下[29]:
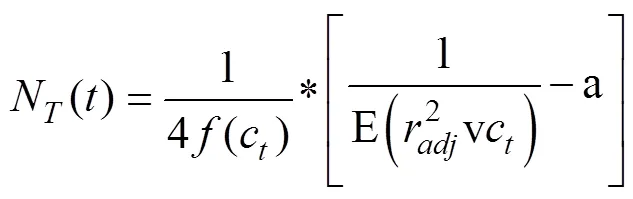
式中N()是代之前的有效群体大小;(c)是用来估计代之前重组率c的映射函数;2代表群体LD的大小,用于调整群体的样本量(2=2– (β)–1,是样本数量;样本为分相数据时:β = 2,样本数据未分相时:β = 1);a是用于修正突变值的常数。
1.1.4 基于群体参数模拟
近似贝叶斯(approximate Bayesian computation, ABC)算法通过数据模拟的方式,逼近似然函数的效果,适用于复杂模型中似然函数推断困难或无法计算的情况[30]。该方法先根据观测数据相关遗传参数(杂合度、遗传距离等)确定先验分布,再对先验分布经过抽样得到待选参数并带入模型进行模拟,随后比较观测数据与模拟数据的差异,如果小于设定的阈值,则接受该待选参数作为后验分布的采样。经过多次采样、仿真和比较以后,即可得到参数的后验分布。根据贝叶斯法则,在观测数据集中,未知参数的后验概率表达式为:

表示在数据集中的后验概率分布,表示关于的似然函数,表示的先验概率分布。表示边缘概率分布,通常忽略不计,故上式可以写为:

ABCtoolbox[31]是基于ABC方法软件,其能够执行完整ABC分析的所有步骤,从先验分布的遗传参数采样、数据模拟,到汇总计算后验分布及概率,再到结果可视化。该软件常结合Fastsimcoal2[25]的数据模拟功能进行优化。为了更加精准地估计群体的演化历史,Sanchez等[32]基于深度学习中的人工神经网络(artificial neural networks, ANN)方法设计了MLP (multi-layer perceptron)、Custom CNN (convolutional neural network)和SPIDNA (sequence position informed deep neural architecture)等架构,利用群体中样本个体的SNP数据来推断有效历史群体大小,其中SPIDNA的估计效果最佳;并且该研究还发现将深度学习与ABC方法结合起来能够对结果进一步优化。
1.2 选择信号研究及检测方法
自然及人工选择是驯化过程中关键的影响因素,经过长期定向的高强度选择,让满足人类需求的特定性状固定下来。选择导致目标基因座,以及相邻基因座中等位基因的频率发生变化[33],并在基因组中留下相应的印记。未被选择的等位基因及其连锁基因的频率降低的现象被称为“选择性清扫”(selective sweep)[34]。而目标基因座临近区域中等位基因频率的变化模式与中性理论预期的不同,被称为“搭便车效应”(hitchhiking)[35]。两者是选择信号的不同表述方式,而所体现的群体遗传事件的背景相同。选择信号在基因组上所产生印迹的主要特征为:(1)被选择位点极端高的等位基因频率;(2)被选择区域长范围增加的单倍型纯合度;(3)群体分化。依据所选择的等位基因或单倍型的起始频率和终端频率,选择信号依次被划分为两种:硬选择(hard sweep)和温和选择(soft sweep)[34,36,37]。根据被选择等位基因或单倍型频率方向的变化不同,选择又可分为正向选择(positive selection)、负向选择(negative selection)和平衡选择(balancing selection)[12,34,38,39]。
1.2.1 基于经典群体遗传学特征检测
选择信号的验证统计量大多都针对正向选择和负向选择展开设计[34,40]。随着基因组测序技术的广泛应用,选择信号被更加准确地识别。目前这方面的综述相对较多,本文不做赘述。检测选择信号最常用的方法可以分为五大类[41]:基于序列水平的替换率(substitution rates),如MKT和dN/dS检验;基于LD,主要有LRH (long-range haplotype)、EHH (extended haplotype homozygosity)和iHS (integrated haplotype score)检验;基于等位基因频谱(allele frequency spectrum),包括Tajima’s、Fay以及Wu’s H检验;以及基于群体分化(differences between populations),包括ST、XP-CLR (cross population composite likelihood ratio)、LSBL (locus-specific branch lengths)、XP-EHH (cross population-extended haplotype homozygosity)和hapFLK检验等[42~44];还有一些复合策略方法综合考虑以上特征,主要包括CMS (composite of signals)、Meta-SS、CSS (composite selection signals)和DCMS (de-correlated composite of signals)方法[45~48]。

图1 受到正向选择的等位基因信号与经典统计量检测方法的关系
典型的正向选择过程(图左):1~2:受到环境等外在选择压力后, 基因组中的某中性区域出现了一个新生有益突变(显示为一个黑色等位基因);3:随后携带该突变的个体在种群中的占比迅速攀升,并且位于其周围与其相连锁的中性突变频率也随之增加(“搭便车效应”),形成了一个受选择区间;4:通过个体间的重组进一步增加了该有益突变的频率,最终该有利突变在种群中固定下来,形成了受选择区域。根据文献[49]修改绘制。
1.2.2 基于机器学习检测
随着基因组数据的爆炸式增长,如何在庞大的数据中提取有利信息并得到真实可信的结论成为现今研究者的一大挑战。Schrider等[50]研究认为,经典统计学方法难以满足群体遗传学目前的发展,功能更强大的机器学习方法才是大数据时代的有利工具。机器学习目前可分为3大类别:无监督学习、有监督学习和半监督学习[12]。研究表明,有监督学习更适合检测自然选择信号,且相比于传统统计量方法有着更优的表现。有监督学习[51]算法包括支持向量机(support vector machine, SVM)、决策树(decision tree)、随机森林(random forest)、神经网络(neural network)等[12]。Lin等[52]通过组合学习将H、π、W、Tajima’s、iHH和Fay & Wu’s H等几个传统统计量综合起来训练,其优势在于能够准确的将自然选择信号和群体历史事件辨别开来。群体大小变化、自然和人工选择等事件可能同时发生,并在基因组中留下类似的印记。如果忽略之间的相互影响,或者不进行细致的剖分,估计出来的遗传参数可能存在偏差,导致做出错误推断。这一方法运用到了两个分类器(classifier):第一个分类器判断用于检测的数据是否存在可能的选择信号,而第二个分类器则是判断上一个分类器检测的信号是源于自然选择还是瓶颈效应。该方法依据Boosting得到的权重高低,判断不同情况下各种统计量在检测信号时的适用范畴[12]。结果表明,iHH在检测近期产生的自然选择时结果更佳;而在检测时间较早期的自然选择时,π更加有效果;对于区分自然选择和瓶颈效应信号时,则W更为擅长[12]。
2 基因选择初始时间估计
除了对驯化过程中选择信号的定性和定量研究,对选择发生时间的研究也逐渐成为研究者感兴趣的问题。虽然用于检测选择信号的方法较多,但是估计选择初始时间的方法仍然有限。现有的估计方法分为两种,其一是针对时间序列数据的选择初始时间估计,该数据要求较多历史时间节点的采集样本,通过综合各时间节点的数据估计某个有利突变的选择初始时间[53]。其中最为典型的方法是 Malaspinas等[54]发表的基于马氏链模型对选择初始时间进行的最大似然估计。第二种方法则是针对非时间序列数据的有利突变选择初始时间估计,目前以近似贝叶斯(ABC)算法最为知名。
2003年,Przeworski等[55]首次运用ABC算法推测有利突变的选择初始时间。作者使用溯祖模拟产生相关模拟数据,再分别利用分离位点数(the number of segregating sites,)、Tajima’s、SFS和单倍型种类数(the number of distinct haplotypes in the sample,)等统计量对这些模拟数据进行分析。作者发现随着统计量数目的增加,选择初始时间估计值的后验概率分布曲线斜率逐渐增大,即估计时间结果更为准确[53]。为了了解生活在美国白沙国家公园的两种蜥蜴(和)由于环境变化而产生适应的遗传机制,Laurent等[56]使用ABC算法结合21种统计量对与肤色相关的基因进行了选择初始时间估计,结果发现群体中的估计时间较早(约1200年前),而在群体中的估计时间则稍晚(约900年前)。虽然两者的估计时间不一致,但其肤色都是在白沙形成之后(约7000年前)改变的。
3 基因交流检测及时间估计
随着对基因交流事件研究的日益深入,其越来越被认为是进化中的一个核心过程。遗传上有别的种群彼此相遇并发生杂交,这一过程广泛影响着驯化动物以及人类的演化进程[57]。
之前的系统发育分析方法大多假定生物类群的分化方式是树状结构,即系统树是个二岐(bifurcating)分支树。然而,随着研究的不断深入,研究人员发现树状结构不能准确的展现真实进化过程。近年来,有研究者提出了更能体现实际进化过程的系统发育网络模式(phylogenetic network),以表现多个系统树之间的网状进化结构[58]。网络状进化模式同样适用于驯化事件。驯化被认为是一个长期的、动态的过程[59],涉及野生和家养种群之间的基因交流(驯化过程中以及驯化后)[60]。当来自两个或多个不同种群的个体开始交换遗传物质后,遗传重组将亲本基因组打断成不同大小的片段,来自不同祖先亚群的染色体片段在子代染色体上镶嵌混合[61]。基因交流可以是单次事件,也可能连续发生。随着世代数增加重组事件也增多,LD片段逐渐变短。每个祖先亲本种群的单倍型片段的期望长度是自初始交流事件产生以来的世代数的函数。通过比较祖先个体或群体的基因组,可以推断遗传自某一祖先群体的基因组片段的构成和比例,即局部和全局祖源分析。基因交流或基因渗入(introgression)是普遍的生物学过程,近年来受到广泛的关注。一个群体可以通过基因交流快速获得新的优势等位基因,增加群体的适应性。通过基因交流获得的基因可能帮助受体获得某些新的功能或者适应环境的变化,这种渗入称为适应性基因渗入(adaptive introgression)[62]。2015年,艾华水等[63]研究发现中国南北(低高纬度)猪种()X染色体上存在两种长达14 Mb的低重组单倍型[64],该研究应用分子钟模型估算该14 Mb差异单倍型在约8.5百万年前分化,远早于本地猪的分化时间;推测可能由于一种已经灭绝的对中国北方猪X染色体上渗入的基因帮助中国北方猪适应了寒冷的环境。
检测是否发生基因交流也是驯化研究的主要内容之一,目前主流的方法主要有f4和f3统计量[65,66]、统计量[67]和基于统计量衍生出来的DFOIL方法[68]等。一些分析方法如主成分分析(principal components analysis, PCA)、Admixture、Structure等工具可用来提示基因渗入的发生。Reich等[66]提出的f4统计量,通过衡量4个群体的系统发生关系与群体间等位基因频率差异之间的关系来判断可能的基因渗入。对于群体A、B、C和D,以及假定的总体拓扑(A,B)(C,D),f4统计量为A和B之间以及C和D之间等位基因频率差的乘积,即:(a-b)(c-d)。如果群体间没有混杂,f4统计量的期望值为0。如果f4统计量的观测值显著偏离0,则提示至少有一个群体存在混杂。f4统计量还有一个简单版本f3统计量,用于检测群体A是否由群体B和群体C混杂而来,即:(a-b)(a-c)。当f3统计量为负时,提示群体A由群体B和群体C混杂而来。
统计量也是检测基因交流的主流方法之一[69]。统计量中定义了H1与H2两个姐妹群体,H3为渗入源群体,H4为外群,并将外群等位基因认定为祖先等位基因。统计量只能用于双等位基因位点,其中定义a为祖先等位基因,b为衍生等位基因。统计量的公式为(修改自文献[69]):

如果没有基因交流发生,统计量的期望值则为0。而当统计量为正值时,结果表明H2群体与H3群体共享更多位点,则推断H3对H2有基因渗入。相反的,当统计量为负值,则表明H1群体与H3群体共享更多位点,即推断H3对H1群体有基因渗入。统计量的设计原理需要渗入源和被渗入群体在等位基因状态上差异较大,否则统计量将不会检测到基因渗入信号[69]。统计量最早用于检测尼安德特人()对现代人祖先的基因渗入,后广泛应用于动物驯化分析。Frantz等[70]使用统计量对东南亚野猪()与家猪之间的基因流事件进行了研究,证明了东南亚桑德兰群岛上的物种之间存在交流事件,且共享着大量的等位基因。
对基因交流时间进行估计是基因交流鉴定分析的扩展,可以帮助更好的理解复杂的群体演化历程。子代个体的基因组包含了从不同祖先而来的片段,由于重组事件打断了染色体片段[71~73],利用位点间LD和单倍型片段在后代个体基因组中的分布这两个原理可推断交流事件发生的日期[74]。
近年来,已经开发了多个估计交流事件时间的工具[75]。(表1,表2) Patterson等[65]于2012年发布了包含ROLLOFF在内等多个基因交流分析方法的软件包Admixtools,其中ROLLOFF是最早基于两个位点之间的连锁不平衡信息来研究基因交流时间的方法[76]。该方法认为通过基因交流产生的LD随着重组的发生呈指数衰减(e–nd,为自交流以来的世代数,为SNP之间的遗传距离)。ROLLOFF计算成对标记之间的LD统计量与反映祖先群体中等位基因频率差异权重之间的相关系数,分析标记间遗传距离的增加对相关系数的影响,并利用最小二乘模型对相关系数的衰减进行了指数拟合,最后得到交流事件发生时间的估计值[76]。

表1 基于LD估计交流时间工具汇总
根据文献[75]修改总结。

表2 基于单倍型/祖先区块大小分布估计交流时间工具汇总
根据文献[75]修改总结。
根据模拟及真实数据的测试,ROLLOFF可以对过去500代内发生的交流事件的时间进行准确的估计。但是,对于小样本量、交流比例低和交流时间较早群体的估计中会产生轻微的向上偏差。同时该方法还会受瓶颈事件和基因漂变等影响,但是几乎不受未校正祖先群体的影响[76]。ALDER[77]拓展了ROLLOFF的方法,利用由混合片段引起的连锁不平衡的指数衰减作为遗传距离函数。ALDER提出了一个新的加权LD统计量,该统计量可用于推断群体的混合比例和交流时间,相比之前方法其对于参考群体的约束更少。MALDER[77]则是ALDER的一个拓展版本,其可以运用于多个交流事件同时发生的情形,能够反应更真实的历史事件[75]。
Chimusa等[75]通过利用模拟数据与真实数据的对比发现MALDER和GLOBETROTTER两种工具的精度最佳。其中GLOBETROTTER[81]考虑到群体迁徙、交流事件可能多次发生和涉及多个群体的情况。该方法将样本中的每个个体视为接收者,其染色体是其他个体贡献的DNA片段重建而来。首先使用工具CHROMOPAINTER[84]将混合个体的染色体分解为各个区块,并将各区块与最可能的祖先个体相关联,然后确定交流种群的完整单倍型区段并将其与不同的祖先种群配对。该方法对于每组拟合一条指数衰减分布曲线并估计最佳拟合率,然后估计其发生的时间。Galaverni等[85]对意大利狼(L)群体使用PCADMIX工具结合ALDER计算群体的混合时间,其结果表明基因交流大多数在采样前的3到4代发生,而最早的事件可追溯到采样之前的19代。
4 群体遗传学用于动物驯化的研究
随着高通量测序及SNP芯片成本逐渐降低,在群体水平研究动物驯化的报道日益增加。目前对于猪、鸡、绵羊()和山羊()等家养动物的研究进展较多。
4.1 家猪
约在10,000年前,猪在近东和中国被驯化。驯化过程中,猪受到自然选择和人工选择的共同作用,在外形、繁殖力、生长发育和环境适应性等方面表现出显著改变[63]。目前在中国家猪中已发现多个与驯化性状相关的基因受到选择。例如, 与神经系统相关的基因与免疫应答相关的基因以及与生长相关的基因等[86]。但是,目前仍缺乏被选择基因选择初始时间的研究报道。梁作翔[53]使用ABC算法估计了部分中国家猪中与驯化性状相关基因的选择压力和选择初始时间,并比较了不同基因的选择初始时间差异。结果表明,与神经系统相关的基因(约8,473.32年前)和与生长相关的基因(约1,587.75年前)之间的选择初始时间相差甚远。经过估计多个基因选择初始时间后, 其认为中国家猪的驯化是一个不断复杂变化的过程, 与同一驯化性状相关的基因既包括早期选择又包括近期选择,持续不同的驯化过程影响着这些性状的改变。最近的研究中,Chen等[87]利用266只欧亚野猪和家猪的全基因组测序数据,得到了法系长白猪(French Large White, FLW)与中国猪之间的渗入比例,并通过ALDER软件计算得到法系长白猪在约200~300年前与华南猪(Southern Chinese pigs, SCN)和华东猪(Eastern Chinese pigs, ECN)发生过基因交流事件。
4.2 家鸡
家鸡驯化于8000~10,000年前,是最早驯化的鸟类之一。在地理隔离和人工选择等因素作用下,家鸡成为表型和用途最为丰富的驯化动物之一,包括给人类提供食物的蛋鸡和肉鸡、用来娱乐打斗的斗鸡(gamecock)及用于观赏消遣的观赏鸡等。Rubin等[88]首次对9个鸡群体进行了全基因组测序(whole genome sequencing, WGS),其中包括红色原鸡(Red Junglefowl)、肉鸡及蛋鸡。根据标准化杂合度(Z- transformations of the pooled heterozygosity, ZP)方法发现基因在驯化中受到了强烈选择。进一步研究发现基因有一个非同义突变发生在外显子区域,可能与鸡季节性发情相关。李明洲课题组利用统计量检测了红色原鸡和10种家鸡之间的基因渗入,发现红色原鸡与藏鸡之间存在更多的渗入区域,可能跟野生红色原鸡与山区散养的藏鸡经常混在一起生活所导致。该研究还发现藏鸡长期生活在高海拔地区的过程中,获得了适应高海拔环境的能力,例如其红细胞数量、血氧亲和力和血红蛋白浓度都有所增加,而通过π、ZST(Z-transformations ofST)和Tajima’s检验等方法进行选择信号分析得到,和基因在参与藏鸡适应高海拔输送氧的方面可能起作用。该研究也发现藏鸡中GTP酶(GTPase)的调控活性显著增加,这表明能量代谢对于维持藏鸡体温的重要性[89]。然而,基因渗入除了发生在野生鸡种和地方鸡种外,商业品种也会有基因渗入到家鸡当中。张春媛等[90]通过比较群体单倍型相似性发现在我国商业鸡种的基因渗入到了地方鸡种基因组中,而由于我国遗传育种保护工作起步较晚,很多方面缺少有效监管,导致基因渗入事件的发生,可能对我国家鸡遗传资源造成了基因污染。
4.3 绵羊和山羊
绵羊和山羊是最早被驯化的反刍动物,源于扎格罗斯山脉附近,已有10,000年的驯化历史[91,92]。作为世界农业经济的重要组成部分,为人们提供了包括肉、毛、奶、皮革等产品[93]。李孟华课题组利用来自129个家羊群体和包括亚洲摩弗伦(Asiatic mouflon)、欧洲摩弗伦(European mouflon)、盘羊(argali)、乌利阿尔羊(urial),大角羊(bighorn)、瘦角羊(thinhorn)和雪羊(snow sheep)等7种野生羊种的3938个样本数据,结合40年间117个气候变化的数据,通过统计量及XP-CLR等方法解析了野生绵羊物种中与抗菌及先天性免疫有关的基因渗入到家养绵羊中,并对抵御肺炎和快速适应气候做出了贡献[94]。姜雨课题组研究了山羊驯化基因的起源,利用来自世界各地的164只家养山羊、24只野山羊、56个山羊化石及6个其他野羊物种的基因组数据进行分析。运用f3统计量和统计量分析了山羊中的基因交流事件,并使用ST和XP-EHH方法进行选择信号分析,推断提高羊群胃肠道抗寄生虫能力的基因使得山羊能够更加适应人类环境,并在山羊驯化过程中起着重要的作用[95]。基于群体基因组学下的遗传变异研究加深了人们对绵羊和山羊遗传及适应机制的认知。
5 结语与展望
群体遗传学用于动物驯化研究,极大地扩展了研究的内容,并推动了学科的发展。一些经典的研究内容,如基因组选择信号和基因交流信号的检测已经相对比较成熟,而对群体历史及时间估计等方面的方法研究可做的工作还有很多。对于群体历史推断,不同的方法各具优点,例如在缺失先验模型的情况下推断种群有效群体大小历史变化和种群间分化时间时,可以利用PSMC、MSMC2及SMC++等方法,不过这类方法没有过多考虑群体之间的基因流事件,其历史真实性会受到影响[96];SFS方法对重构复杂的多群体历史变化和推测群体之间的迁徙事件具有很好的效果;推测近期历史事件则可以利用基于LD或者IBD (identical by descent)的方法来得到较为准确的时间。为了更准确的估算结果则需要充分考虑可能发生的历史事件,构建更全面的模型进行推断。基于恰当模型量化特定群体历史的方式逐渐成为一种主流,然而模型的适合度固然重要,但是其稳定可行性却更为重要。一个模型若要在任何参数上都与真实的群体历史相匹配的概率极其微小,我们需要带着实际科学问题去设计不同的模型来研究我们最感兴趣的问题,通过利用赤池信息量(Akaike information criterion, AIC)或贝叶斯信息量(Bayesian information criterion, BIC)等标准来筛选与实际数据最吻合的模型。
在过去的几十年中,关于驯化的考古学研究和遗传学研究持续开展。研究驯化的时间节点不仅有助于了解驯化对象自身的群体历史,而且有助于了解人类的迁移扩散和文明发展史[7]。对于基因选择初始时间的估计,种群动态历史也是重要的影响因素之一。Ormond等[97]在估计鹿鼠(deer mouse)中某一个与皮肤颜色变化有关的有利突变的选择初始时间时,发现如果在模拟数据中不加以种群动态历史模型推断,则根据真实数据估计所产生的后验概率分布显著离散,即准确度偏低。而在添加了种群动态历史模型之后,估计真实数据的后验概率分布就变得非常集中,即准确率显著提高。对于交流时间估计来说,近年来由于基因分型和测序技术的进步,以及统计和计算工具的发展使得以上方法能够更精确的估计基因交流事件发生的日期。基因交流的研究可以用来揭示很多极为有趣的生物学问题,例如,Frantz等[98]通过对多种模型进行比较展示了十分复杂的猪的驯化进程。野猪和家猪群体之间的基因交流在驯化过程中或驯化后均普遍存在。另外,人工选择又抵消掉野生和家养群体基因交流产生的均一化影响,使得两者之间的形态及行为差异化。但是,对复杂多混合种群基因组的祖先片段分布或精确定位祖先片段并进行建模仍是一个尚未解决的问题,以至于对较早期或多群体混合的复杂交流事件的时间估计还是不够精准[75]。
为了更好的研究动物驯化的历史,应综合运用跨学科的知识,包括群体遗传学、地理地质学、考古学和历史学等进行更全面的研究。在材料方面,优化和改进古代DNA (ancient DNA, aDNA)分离和扩增技术,为古代尤其是史前的数据提供证据支持。另外,基于大量的基因组数据可以发掘出更多群体遗传学特征,用于发现驯化过程中基因组的改变,以便进行更全面的分析。在分析方法上,引入更多的多元统计学和机器学习方法,可以更好的进行分类和定量研究。最后,面对大量测序数据的积累,需加强对于处理超大规模群体数据的算法改进和包括GPU[99]和FPGA[100]在内的硬件加速来满足科学研究的需求。
[1] Pan ZY, He XY, Wang XY, Guo XF, Cao XH, Hu WP, Di R, Liu QY, Chu MX. Selection signature in domesticated animals., 2016, 38(12): 1069–1080.潘章源, 贺小云, 王翔宇, 郭晓飞, 曹晓涵, 胡文萍, 狄冉, 刘秋月, 储明星. 家养动物选择信号研究进展. 遗传, 2016, 38(12): 1069–1080.
[2] Zeder MA. Core questions in domestication research., 2015, 112(11): 3191–3198.
[3] Gamble C, Davies W, Pettitt P, Richards M. Climate change and evolving human diversity in Europe during the Last Glacial., 2004, 359(1442): 243–254.
[4] Fabrice T. Animal domestication: A brief overview. London: IntechOpen. 2019.
[5] Zeder MA. The domestication of animals., 1982, 9(4): 321–327.
[6] Larson G, Piperno DR, Allaby RG, Purugganan MD, Andersson L, Arroyo-Kalin M, Barton L, Climer Vigueira C, Denham T, Dobney K, Doust AN, Gepts P, Gilbert MTP, Gremillion KJ, Lucas L, Lukens L, Marshall FB, Olsen KM, Pires JC, Richerson PJ, de Casas RR, Sanjur OI, Thomas MG, Fuller DQ. Current perspectives and the future of domestication studies., 2014, 111(17): 6139–6146.
[7] Li J, Zhang YP. Advances in research of the origin and domestication of domestic animals., 2009, 17(4): 1–11.李晶, 张亚平. 家养动物的起源与驯化研究进展. 生物多样性, 2009, 17(4): 1–11.
[8] Diamond J. Evolution, consequences and future of plant and animal domestication., 2002, 418(6898): 700–707.
[9] Evershed RP, Payne S, Sherratt AG, Copley MS, Coolidge J, Urem-Kotsu D, Kotsakis K, Ozdoğan M, Ozdoğan AE, Nieuwenhuyse O, Akkermans PMMG, Bailey D, Andeescu RR, Campbell S, Farid S, Hodder I, Yalman N, Ozbaşaran M, Biçakci E, Garfinkel Y, Levy T, Burton MM. Earliest date for milk use in the Near East and southeastern Europe linked to cattle herding., 2008, 455(7212): 528–531.
[10] Outram AK, Stear NA, Bendrey R, Olsen S, Kasparov A, Zaibert V, Thorpe N, Evershed RP. The earliest horse harnessing and milking., 2009, 323(5919): 1332–1335.
[11] Daniel LH, Andrew GC. Principles of population genetics. 4th ed. Sinauer Associates,Sunderland, MA, 2006.
[12] Shi Y, Li HP. Population genomics: from classical statistics to supervised learning., 2019, 49(4): 445–455.施怿, 李海鹏. 群体基因组学方法:从经典统计学到有监督学习. 中国科学: 生命科学, 2019, 49(4): 445– 455.
[13] Zheng ZQ. Population structure and genetic introgression from wild relatives in worldwide goat populations [Dissertation]. Northwest A&F University, 2019.郑竹清. 世界山羊群体遗传结构及其野生近缘种基因渗入研究[学位论文]. 西北农林科技大学, 2019.
[14] Wang GD, Zhai WW, Yang HC, Wang L, Zhong L, Liu YH, Fan RX, Yin TT, Zhu CL, Poyarkov AD, Irwin DM, Hytönen MK, Lohi H, Wu CI, Savolainen P, Zhang YP. Out of southern East Asia: the natural history of domestic dogs across the world., 2016, 26(1): 21–33.
[15] Peters J, Lebrasseur O, Deng H, Larson G. Holocene cultural history of Red jungle fowl () and its domestic descendant in East Asia., 2016, 142: 102–119.
[16] Wang MS, Thakur M, Peng MS, Jiang Y, Frantz LAF, Li M, Zhang JJ, Wang S, Peters J, Otecko NO, Suwannapoom C, Guo X, Zheng ZQ, Esmailizadeh A, Hirimuthugoda NY, Ashari H, Suladari S, Zein MSA, Kusza S, Sohrabi S, Kharrati-Koopaee H, Shen QK, Zeng L, Yang MM, Wu YJ, Yang XY, Lu XM, Jia XZ, Nie QH, Lamont SJ, Lasagna E, Ceccobelli S, Gunwardana HGTN, Senasige TM, Feng SH, Si JF, Zhang H, Jin JQ, Li ML, Liu YH, Chen HM, Ma C, Dai SS, Bhuiyan AKFH, Khan MS, Silva GLLP, Le TT, Mwai OA, Ibrahim MNM, Supple M, Shapiro B, Hanotte O, Zhang GJ, Larson G, Han JL, Wu DD, Zhang YP. 863 genomes reveal the origin and domestication of chicken., 2020, 30(8): 693–701.
[17] Gao F, Li HP. Application of computer simulators in population genetics., 2016, 38(8): 707–717.高峰, 李海鹏. 群体遗传学模拟软件应用现状. 遗传, 2016, 38(8): 707–717.
[18] Li H, Durbin R. Inference of human population history from individual whole-genome sequences., 2011, 475(7357): 493–496.
[19] Schiffels S, Durbin R. Inferring human population size and separation history from multiple genome sequences., 2014, 46(8): 919–925.
[20] Terhorst J, Kamm JA, Song YS. Robust and scalable inference of population history from hundreds of unphased whole genomes., 2017, 49(2): 303– 309.
[21] Qanbari S, Strom TM, Haberer G, Weigend S, Gheyas AA, Turner F, Burt DW, Preisinger R, Gianola D, Simianer H. A high resolution genome-wide scan for significant selective sweeps: an application to pooled sequence data in laying chickens., 2012, 7(11): e49525.
[22] Li HP, Stephan W. Inferring the demographic history and rate of adaptive substitution in., 2006, 2(10): e166.
[23] Wakeley J, Hey J. Estimating ancestral population parameters., 1997, 145(3): 847–855.
[24] Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data., 2009, 5(10): e1000695.
[25] Excoffier L, Dupanloup I, Huerta-Sánchez E, Sousa VC, Foll M. Robust demographic inference from genomic and SNP data., 2013, 9(10): e1003905.
[26] Barbato M, Orozco-terWengel P, Tapio M, Bruford MW. Snep: a tool to estimate trends in recent effective population size trajectories using genome-wide SNP data., 2015, 6: 109.
[27] Wang JL. Estimation of effective population sizes from data on genetic markers., 2005, 360(1459): 1395–1409.
[28] Hill WG. Estimation of effective population size from data on linkage disequilibrium., 1981, 38(3): 209–216.
[29] Corbin LJ, Liu AYH, Bishop SC, Woolliams JA. Estimation of historical effective population size using linkage disequilibria with marker data., 2012, 129(4): 257–270.
[30] Sunnåker M, Busetto AG, Numminen E, Corander J, Foll M, Dessimoz C. Approximate Bayesian computation., 2013, 9(1): e1002803.
[31] Wegmann D, Leuenberger C, Neuenschwander S, Excoffier L. Abctoolbox: A versatile toolkit for approximate Bayesian computations., 2010, 11(116).
[32] Sanchez T, Cury J, Charpiat G, Jay F. Deep learning for population size history inference: design, comparison and combination with approximate Bayesian computation., 2020.
[33] Tiago Do Prado Paim, Patrícia I, Samuel RP, Alexandre RC, Concepta Margaret Mcmanus Pimentel. Detection and evaluation of selection signatures in sheep., 2018, 53(5): 527–539.
[34] Xue ZYY, Song XW, Wu LH, Wang LZ, Cui JA, Sun ZJ, Zhang Z, Ma YL. The identification methods of selection signatures in livestock and its statistical problems., 2018, 49(6): 1099–1107.薛周舣源, 宋显威, 吴林慧, 王露珍, 崔家安, 孙章健, 张政, 马云龙. 畜禽选择信号检测方法及其统计学问题. 畜牧兽医学报, 2018, 49(6): 1099–1107.
[35] Eisenhaber F. Discovering biomolecular mechanisms with computational biology. Springer, Boston, MA, 2006.
[36] Pennings PS, Hermisson J. Soft sweeps II—molecular population genetics of adaptation from recurrent mutation or migration., 2006, 23(5): 1076–1084.
[37] Hermisson J, Pennings PS. Soft sweeps: molecular population genetics of adaptation from standing genetic variation., 2005, 169(4): 2335–2352.
[38] Suzuki Y. Statistical methods for detecting natural selection from genomic data., 2010, 85(6): 359–376.
[39] Nielsen R. Molecular signatures of natural selection., 2005, 39: 197–218.
[40] Lohmueller KE, Bustamante CD, Clark AG. Detecting directional selection in the presence of recent admixture in African-Americans., 2011, 187(3): 823–835.
[41] Wang YZ, Zhao YQ. Research progress of genomic signature of selection and its detection methods., 2019, 40(5): 1–6.王宇占, 赵毅强. 基因组水平的选择信号及其检测方法研究进展. 家畜生态学报, 2019, 40(5): 1–6.
[42] de Simoni Gouveia JJ, da Silva MVGB, Paiva SR, de Oliveira SMP. Identification of selection signatures in livestock species., 2014, 37(2): 330– 342.
[43] Chen H, Patterson N, Reich D. Population differentiation as a test for selective sweeps., 2010, 20(3): 393–402.
[44] Oleksyk TK, Smith MW, O'Brien SJ. Genome-wide scans for footprints of natural selection., 2010, 365(1537): 185–205.
[45] Grossman SR, Shlyakhter I, Karlsson EK, Byrne EH, Morales S, Frieden G, Hostetter E, Angelino E, Garber M, Zuk O, Lander ES, Schaffner SF, Sabeti PC. A composite of multiple signals distinguishes causal variants in regions of positive selection., 2010, 327(5967): 883–886.
[46] Utsunomiya YT, Pérez O'Brien AM, Sonstegard TS, Van Tassell CP, do Carmo AS, Mészáros G, Sölkner J, Garcia JF. Detecting loci under recent positive selection in dairy and beef cattle by combining different genome- wide scan methods., 2013, 8(5): e64280.
[47] Randhawa IAS, Khatkar MS, Thomson PC, Raadsma HW. Composite selection signals can localize the trait specific genomic regions in multi-breed populations of cattle and sheep., 2014, 15: 34.
[48] Ma Y, Ding X, Qanbari S, Weigend S, Zhang Q, Simianer H. Properties of different selection signature statistics and a new strategy for combining them., 2015, 115(5): 426–436.
[49] Biswas S, Akey JM. Genomic insights into positive selection., 2006, 22(8): 437–446.
[50] Schrider DR, Kern AD. Supervised machine learning for population genetics: a new paradigm., 2018, 34(4): 301–312.
[51] Kotsiantis SB. Supervised machine learning: a review of classification techniques., 2007, 31: 3–24.
[52] Lin K, Li HP, Schlötterer C, Futschik A. Distinguishing positive selection from neutral evolution: boosting the performance of summary statistics., 2011, 187(1): 229–244.
[53] Liang ZX. Analysis of pig domestication and ancestry using genomewide SNP information[Dissertation]. China Agricultural University, 2019.梁作翔. 利用全基因组SNP信息研究家猪的驯化及祖先来源[学位论文]. 中国农业大学, 2019.
[54] Malaspinas AS, Malaspinas O, Evans SN, Slatkin M. Estimating allele age and selection coefficient from time-serial data., 2012, 192(2): 599–607.
[55] Przeworski M. Estimating the time since the fixation of a beneficial allele., 2003, 164(4): 1667–1676.
[56] Laurent S, Pfeifer SP, Settles ML, Hunter SS, Hardwick KM, Ormond L, Sousa VC, Jensen JD, Rosenblum EB. The population genomics of rapid adaptation: disentangling signatures of selection and demography in white sands lizards., 2016, 25(1): 306–323.
[57] Medina P, Thornlow B, Nielsen R, Corbett-Detig R. Estimating the timing of multiple admixture pulses during local ancestry inference., 2018, 210(3): 1089–1107.
[58] Cheng CH, Huang Y. Construction and application of phylogenetic network., 2008, 30(3): 215–221.程春花, 黄原. 系统发育网络的构建与应用. 昆虫分类学报, 2008, 30(3): 215–221.
[59] Dobney K, Larson G. Genetics and animal domestication: New windows on an elusive process., 2006, 269(2): 261–271.
[60] Marshall FB, Dobney K, Denham T, Capriles JM. Evaluating the roles of directed breeding and gene flow in animal domestication., 2014, 111(17): 6153–6158.
[61] Winkler CA, Nelson GW, Smith MW. Smith. Admixture mapping comes of age., 2010, 11: 65–89.
[62] Racimo F, Sankararaman S, Nielsen R, Huerta-Sánchez E. Evidence for archaic adaptive introgression in humans., 2015, 16(6): 359–371.
[63] Ai HS, Fang XD, Yang B, Huang ZY, Chen H, Mao LK, Zhang F, Zhang L, Cui LL, He WM, Yang J, Yao XM, Zhou LS, Han LJ, Li J, Sun SL, Xie XH, Lai BX, Su Y, Lu Y, Yang H, Huang T, Deng WJ, Nielsen R, Ren J, Huang LS. Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing., 2015, 47(3): 217–225.
[64] Liang SY, Zhou ZK, Hou SS. The research progress of farm animal genomics based on sequencing technologies., 2017, 39(4): 276–292.梁素芸, 周正奎, 侯水生. 基于测序技术的畜禽基因组学研究进展. 遗传, 2017, 39(4): 276–292.
[65] Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan YP, Genschoreck T, Webster T, Reich D. Ancient admixture in human history., 2012, 192(3): 1065–1093.
[66] Reich D, Thangaraj K, Patterson N, Price AL, Singh L. Reconstructing Indian population history., 2009, 461(7263): 489–494.
[67] Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai WW, Fritz MHY, Hansen NF, Durand EY, Malaspinas AS, Jensen JD, Marques-Bonet T, Alkan C, Prüfer K, Meyer M, Burbano HA, Good JM, Schultz R, Aximu-Petri A, Butthof A, Höber B, Höffner B, Siegemund M, Weihmann A, Nusbaum C, Lander ES, Russ C, Novod N, Affourtit J, Egholm M, Verna C, Rudan P, Brajkovic D, Kucan Ž, Gušic I, Doronichev VB, Golovanova LV, Lalueza-Fox C, de la Rasilla M, Fortea J, Rosas A, Schmitz RW, Johnson PLF, Eichler EE, Falush D, Birney E, Mullikin JC, Slatkin M, Nielsen R, Kelso J, Lachmann M, Reich D, Pääbo S. A draft sequence of the Neandertal genome., 2010, 328(5979): 710– 722.
[68] Pease JB, Hahn MW. Detection and polarization of introgression in a five-taxon phylogeny., 2015, 64(4): 651–662.
[69] Zheng YC, Janke A. Gene flow analysis method, the-statistic, is robust in a wide parameter space., 2018, 19(1): 10.
[70] Frantz LAF, Schraiber JG, Madsen O, Megens HJ, Bosse M, Paudel Y, Semiadi G, Meijaard E, Li N, Crooijmans RPMA, Archibald AL, Slatkin M, Schook LB, Larson G, Groenen MAM. Genome sequencing reveals fine scale diversification and reticulation history during speciation in., 2013, 14(9): R107.
[71] Sanderson J, Sudoyo H, Karafet TM, Hammer MF, Cox MP. Reconstructing past admixture processes from local genomic ancestry using wavelet transformation., 2015, 200(2): 469–481.
[72] Pugach I, Matveyev R, Wollstein A, Kayser M, Stoneking M. Dating the age of admixturewavelet transform analysis of genome-wide data., 2011, 12(2): R19.
[73] Xu SH, Huang W, Qian J, Jin L. Analysis of genomic admixture in Uyghur and its implication in mapping strategy., 2008, 82(4): 883–894.
[74] Price AL, Tandon A, Patterson N, Barnes KC, Rafaels N, Ruczinski I, Beaty TH, Mathias R, Reich D, Myers S. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations., 2009, 5(6): e1000519.
[75] Chimusa ER, Defo J, Thami PK, Awany D, Mulisa DD, Allali I, Ghazal H, Moussa A, Mazandu GK. Dating admixture events is unsolved problem in multi-way admixed populations., 2020, 21(2): 144–155.
[76] Moorjani P, Patterson N, Hirschhorn JN, Keinan A, Hao L, Atzmon G, Burns E, Ostrer H, Price AL, Reich D. The history of African gene flow into Southern Europeans, Levantines, and Jews., 2011, 7(4): e1001373.
[77] Loh PR, Lipson M, Patterson N, Moorjani P, Pickrell JK, Reich D, Berger B. Inferring admixture histories of human populations using linkage disequilibrium., 2013, 193(4): 1233–1254.
[78] Zhou Y, Qiu HX, Xu SH. Modeling continuous admixture using admixture-induced linkage disequilibrium., 2017, 7: 43054.
[79] Zhou Y, Yuan K, Yu Y, Ni X, Xie P, Xing EP, Xu S. Inference of multiple-wave population admixture by modeling decay of linkage disequilibrium with polynomial functions., 2017, 118(5): 503–510.
[80] Ni XM, Yuan K, Yang X, Feng QD, Guo W, Ma ZM, Xu SH. Inference of multiple-wave admixtures by length distribution of ancestral tracks., 2018, 121(1): 52–63.
[81] Hellenthal G, Busby GBJ, Band G, Wilson JF, Capelli C, Falush D, Myers S. A genetic atlas of human admixture history., 2014, 343(6172): 747–751.
[82] Ni XM, Yang X, Guo W, Yuan K, Zhou Y, Ma ZM, Xu SH. Length distribution of ancestral tracks under a general admixture model and its applications in population history inference., 2016, 6: 20048.
[83] Corbett-Detig R, Nielsen R. A hidden Markov model approach for simultaneously estimating local ancestry and admixture time using next generation sequence data in samples of arbitrary ploidy., 2017, 13(1): e1006529.
[84] Lawson DJ, Hellenthal G, Myers S, Falush D. Inference of population structure using dense haplotype data., 2012, 8(1): e1002453.
[85] Galaverni M, Caniglia R, Pagani L, Fabbri E, Boattini A, Randi E. Disentangling timing of admixture, patterns of introgression, and phenotypic indicators in a hybridizing wolf population., 2017, 34(9): 2324– 2339.
[86] Zhu YL, Li WB, Yang B, Zhang ZY, Ai HS, Ren J, Huang LS. Signatures of selection and interspecies introgression in the genome of Chinese domestic pigs., 2017, 9(10): 2592–2603.
[87] Chen H, Huang M, Yang B, Wu ZP, Deng Z, Hou Y, Ren J, Huang LS. Introgression of Eastern Chinese and Southern Chinese haplotypes contributes to the improvement of fertility and immunity in European modern pigs., 2020, 9(3): giaa014.
[88] Rubin CJ, Zody MC, Eriksson J, Meadows JRS, Sherwood E, Webster MT, Jiang L, Ingman M, Sharpe T, Ka S, Hallböök F, Besnier F, Carlborg O, Bed'hom B, Tixier-Boichard M, Jensen P, Siegel P, Lindblad-Toh K, Andersson L. Whole-genome resequencing reveals loci under selection during chicken domestication., 2010, 464(7288): 587–591.
[89] Li DY, Li Y, Li M, Che TD, Tian SL, Chen BL, Zhou XM, Zhang GL, Gaur U, Luo MJ, Tian K, He MN, He S, Xu ZX, Jin L, Tang QZ, Dai YF, Xu HL, Hu YD, Zhao XL, Yin HD, Wang Y, Zhou RJ, Yang CW, Du HR, Jiang XS, Zhu Q, Li MZ. Population genomics identifies patterns of genetic diversity and selection in chicken., 2019, 20(1): 263.
[90] Zhang CY, Lin D, Wang YZ, Peng DZ, Li HF, Fei J, Chen KW, Yang N, Hu XX, Zhao YQ, Li N. Widespread introgression in Chinese indigenous chicken breeds from commercial broiler., 2019, 12(3): 610– 621.
[91] Zeder MA, Hesse B. The initial domestication of goats () in the Zagros Mountains 10, 000 years ago., 2000, 287(5461): 2254–2257.
[92] Wang FH, Zhang L, Li XK, Fan YX, Qiao X, Gong G, Yan XC, Zhang LT, Wang ZY, Wang RJ, Liu ZH, Wang ZX, He LB, Zhang YJ, Li JQ, Zhao YH, Su R. Progress in goat genome studies., 2019, 41(10): 928–938.王凤红, 张磊, 李晓凯, 范一星, 乔贤, 龚高, 严晓春, 张令天, 王志英, 王瑞军, 刘志红, 王志新, 何利兵, 张燕军, 李金泉, 赵艳红, 苏蕊. 山羊基因组研究进展. 遗传, 2019, 41(10): 928–938.
[93] Li XK, Wang G, Qiao X, Fan Y, Zhang L, Ma YH, Nie RX, Wang RJ, He LB, Su R. Research progress on whole-genome sequencing on important domesticated animals., 2018, 34(6): 11–21.李晓凯, 王贵, 乔贤, 范一星, 张磊, 马宇浩, 聂瑞雪, 王瑞军, 何利兵, 苏蕊. 全基因组测序在重要家畜上的研究进展. 生物技术通报, 2018, 34(6): 11–21.
[94] Cao YH, Xu SS, Shen M, Chen ZH, Gao L, Lv FH, Xie XL, Wang XH, Yang H, Liu CB, Zhou P, Wan PC, Zhang YS, Yang JQ, Pi WH, Eer H, Berry DP, Barbato M, Esmailizadeh A, Nosrati M, Salehian-Dehkordi H, Dehghani-Qanatqestani M, Dotsev AV, Deniskova TE, Zinovieva NA, Brem G, Štěpánek O, Ciani E, Weimann C, Erhardt G, Mwacharo JM, Ahbara A, Han JL, Hanotte O, Miller JM, Sim Z, Coltman D, Kantanen J, Bruford MW, Lenstra JA, Kijas J, Li MH. Historical introgression from wild relatives enhanced climatic adaptation and resistance to pneumonia in sheep., 2020, 17: msaa236.
[95] Zheng ZQ, Wang XH, Li M, Li YJ, Yang ZR, Wang XL, Pan XY, Gong M, Zhang Y, Guo YW, Wang Y, Liu J, Cai YD, Chen QM, Okpeku M, Colli L, Cai DW, Wang K, Huang SS, Sonstegard TS, Esmailizadeh A, Zhang WG, Zhang TT, Xu YB, Xu NY, Yang Y, Han JL, Chen L, Lesur J, Daly KG, Bradley DG, Heller R, Zhang GJ, Wang W, Chen YL, Jiang Y. The origin of domestication genes in goats., 2020, 6(21): eaaz5216.
[96] Terhorst J, Kamm JA, Song YS. Robust and scalable inference of population history from hundreds of unphased whole genomes., 2017, 49(2): 303–309.
[97] Ormond L, Foll M, Ewing GB, Pfeifer SP, Jensen JD. Inferring the age of a fixed beneficial allele., 2016, 25(1): 157–169.
[98] Frantz LA, Schraiber JG, Madsen O, Megens HJ, Cagan A, Bosse M, Paudel Y, Crooijmans RPMA, Larson G, Groenen MAM. Evidence of long-term gene flow and selection during domestication from analyses of Eurasian wild and domestic pig genomes., 2015, 47(10): 1141–1148.
[99] Freudenthal JA, Ankenbrand MJ, Grimm DG, Korte A. GWAS-Flow: A GPU accelerated framework for efficient permutation based genome-wide association studies., 2019, 1: 783100.
[100] Bu LN, Wang Q, Gu WJ, Yang RF, Zhu D, Song Z, Liu XJ, Zhao YQ. Improving read alignment through the generation of alternative referenceiterative strategy., 2020, 10(1): 18712.
Progress on animal domestication under population genetics
Zilong Wen, Yiqiang Zhao
Animal domestication is the process of changing wild animals into domesticated animals that can be kept stably for a long period of time. As the content of the Neolithic agricultural revolution, domestication is one of the important milestones of the progress of human civilization. Due to the close relationship between humans and animals, domestication has not only changed the wild state of animals, but also changed the habits and historical processes of human beings. The key question on animal domestication research include who is the ancestors of the domesticated animals were, the changes produced by domestication, and the time and place of domestication. Due to the advances in high-throughput genomic technologies and correspondence analysis methods, animal domestication is generally studied at the population level. Here we discuss the research content of animal domestication under population genetics, including population history, selection signals, as well as gene introgression, and we highlight two new expand contents, namely, dating the initial time of gene selection and the time of gene introgression. Finally, we summarize the recent research progress of major domesticated including pig, chicken, sheep and goat. These advances provide a new insights and perspective for the research on the animal domestication.
domestication; domesticated animals; population genetics; initial time of gene selection; time of gene introgression
2020-08-26;
2020-12-07
转基因重大专项(编号:2018ZX08007001)资助[Supported by National Special Foundation for Transgenic Species of China (No. 2018ZX08007001)]
文子龙,在读硕士研究生,专业方向:生物信息学。E-mail: zilongwen@cau.edu.cn
赵毅强,博士,副教授,博士生导师,研究方向:生物信息学。E-mail: yiqiangz@cau.edu.cn
10.16288/j.yczz.20-268
2021/2/3 10:41:51
URI: https://kns.cnki.net/kcms/detail/11.1913.r.20210202.1002.002.html
(责任编委: 李海鹏)
猜你喜欢
今日农业(2021年11期)2021-08-13
智慧健康(2021年17期)2021-07-30
国际检验医学杂志(2021年7期)2021-04-15
科学大众(2020年10期)2020-07-24
中国产前诊断杂志(电子版)(2020年1期)2020-05-21
当代陕西(2019年6期)2019-04-17
现代检验医学杂志(2016年5期)2016-08-20
遗传(2014年3期)2014-02-28
世界科学(2014年8期)2014-02-28
世界科学(2013年6期)2013-03-11
