
三-(五氟苯基)咔咯锰配合物与DNA碱基的相互作用理论研究
2021-03-09 10:15徐志广
华南师范大学学报(自然科学版) 2021年1期
李 皎, 徐 艳, 许 旋, 徐志广*
(1. 华南师范大学化学学院∥教育部环境理论化学重点实验室, 广州 510006; 2. 珠峰实验学校, 珠海 519000)
DNA是自然界中许多生命有机体内的重要抗肿瘤药物靶标分子,小分子和DNA作用可能会损伤DNA,影响相关蛋白质的合成和翻译,若与肿瘤细胞的DNA作用,可抑制肿瘤细胞的增殖,从而起到抗癌作用[1-2]. 小分子化合物与DNA的相互作用及其核酸酶活性一直是生物化学的重要研究课题之一[3-5]. 小分子和DNA的结合模式主要有2种:共价结合、非共价结合. 一般认为咔咯等卟啉类化合物与DNA的相互作用模式是非共价结合,主要分为4种类型[6]:插入结合、静电作用、沟槽结合和外部结合,其中插入结合和外部结合是咔咯和DNA相互作用中最常见的结合模式[7-8].
有关卟啉化合物及其衍生物与DNA相互作用的研究已有大量报道[9-13],但作为卟啉类似物的芳香大环化合物,咔咯和DNA相互作用的研究相对较少. 近些年,科研人员不断合成出高氧化态的金属咔咯,并在实验中发现金属咔咯能够与DNA相互作用、催化断裂DNA,在光动力治疗和抗肿瘤等生物医学应用方面具有良好前景[14]. 咔咯金属配合物和DNA的相互作用及其核酸酶活性逐渐成为生物化学领域的热点研究之一[15-17]. 咔咯锰配合物的活性中心Mn原子具有良好的氧化还原特性,在结合DNA、催化氧化DNA断裂等方面表现出较好的化学生物活性. 为了从分子水平深入探究咔咯锰配合物和DNA的相互作用机制及构效关系,本文对(TPFC)Mn(Ⅲ)与DNA的碱基以及碱基对的轴向配位性质进行理论计算,为实验研究提供新的理论依据.
1 计算方法
DNA的双链结构非常复杂,因此本文将DNA双链结构分解,选取DNA的4个碱基片段:腺嘌呤(Adenine,A)、鸟嘌呤(Guanine,G)、胞嘧啶(Cytosine,C)、胸腺嘧啶(Thymine,T),对(TPFC)Mn(Ⅲ)与4种碱基以及A=T、C≡G碱基对的轴向配位性质进行理论研究,均以1∶1的数量比进行配位. 实验研究发现,金属咔咯与DNA的主要结合模式为插入结合和外部结合,因此设计了10种(TPFC)Mn(Ⅲ)与A、T、C、G碱基轴向配位的分子模型以及12种(TPFC)Mn(Ⅲ)与A=T、C≡G碱基对轴向配位的分子模型,主要涉及插入结合和外部结合2种结合模式.
本课题组的前期研究[18-19]对(TPFC)Mn(Ⅲ)的理论计算发现其基态是五重态,结构最稳定,故本文研究的所有分子模型均在五重态下进行全优化计算,所有配合物均采用DFT/B3LYP方法进行计算,其中非金属元素(C、N、F、H、O)采用6-31G**基组,金属元素(Mn)采用LanL2DZ 基组[20],分子的NPA电荷、韦伯键级(Wiberg bond Index,WI)、二级微扰稳定化能等均采用Gaussian 03(Revision D.01版)的NBO[21]程序计算. 所有分子的计算过程均采用Gaussian 03程序[22].
2 结果与讨论
2.1 (TPFC)Mn(Ⅲ)和碱基的轴向配位性质
DNA的4种碱基结构以及与(TPFC)Mn(Ⅲ)的轴向配位模型见图1,其中,以G的2个N原子(1、2号)及O原子(3号)、A的3个N原子(1、2、3号)、C的N原子(1号)和O原子(2号)、T的2个O原子(1、2号)作为配位原子和(TPFC)Mn(Ⅲ)配位. 根据每个碱基的配位原子编号,将本文所设计的A、C、G、T和(TPFC)Mn(Ⅲ)轴向配位的10个模型分子,依次命名为GN1、GN2、GO3、AN1、AN2、AN3、CN1、CO2、TO1和TO2(优化几何结构见图2),各个配合物的频率计算均无虚频,结构稳定,对应的几何参数见表1.

图1 A、C、G、T和(TPFC)Mn(Ⅲ)(L)的分子结构
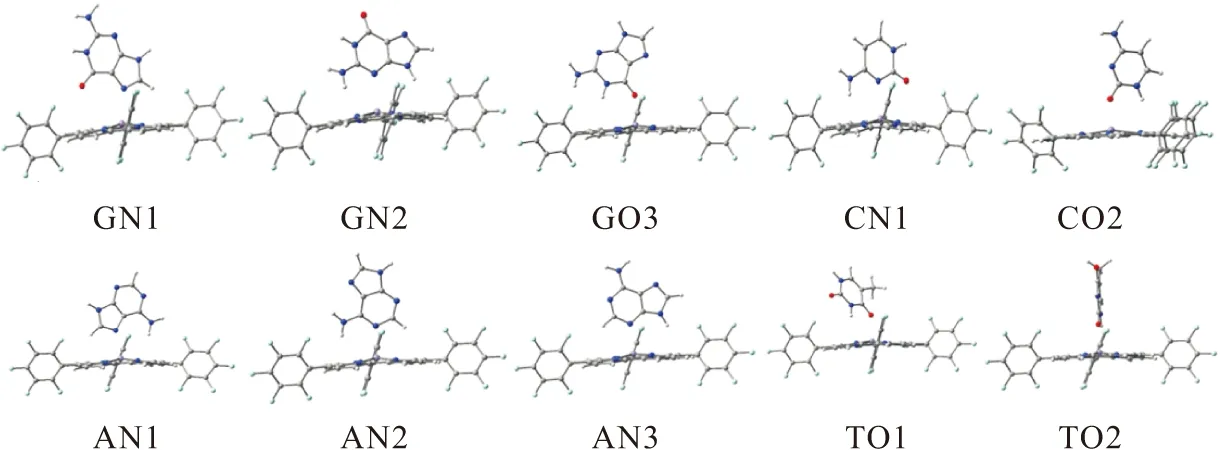
图2 (TPFC)Mn(Ⅲ)(L)的优化几何结构

表1 (TPFC)Mn(Ⅲ)(L)的部分几何结构参数和结合能Table 1 The selected geometrical parameters and binding energy of (TPFC)Mn(Ⅲ)(L)
Mn偏离咔咯平面环的程度越大,越向碱基靠近,(TPFC)Mn(Ⅲ)和碱基的配位结合越强,dMn-4N由0.032 7 nm(CO2)降至0.024 4 nm(GN2);配位键键长dMn-L越短,结合能ΔE的绝对值越大,配合物的结构越稳定,碱基和(TPFC)Mn(Ⅲ)的配位能力越强,配位键键长dMn-L从0.217 3 nm(CO2)增至的0.247 2 nm(GN2),ΔE从-68.39 kJ/mol(CO2)降至的-36.17 kJ/mol(GN2);因此在(TPFC)Mn(Ⅲ)(L)中,配位能力由大到小为:CO2、GO3、TO2、TO1、AN3、AN2、AN1、GN1、CN1、GN2,且碱基上的O原子和Mn原子的结合能力更强.
电荷转移是化学反应的基本过程,根据自然电荷布局分析(表2),ΔQL为碱基与(TPFC)Mn(Ⅲ)配位前后电荷的变化量(碱基与(TPFC)Mn(Ⅲ)配位前电荷为0),ΔQL越大,碱基向(TPFC)Mn(Ⅲ)转移的电子就越多,碱基与(TPFC)Mn(Ⅲ)的配位作用越强,因此配位能力由大到小为:CO2、TO2(或CN1)、GO3、GN1、AN3、AN2、AN1、TO1、GN2,与前面所得结论不一致. 为了深入探讨以上结论不一致的原因,引入韦伯键级WIMn-L作为辅助依据(表2)判断碱基和(TPFC)Mn(Ⅲ)之间的配位性质,WIMn-L越大,碱基和(TPFC)Mn(Ⅲ)的配位作用越强. 由此可判断(TPFC)Mn(Ⅲ)和碱基之间的结合能力由大到小依次为:CO2、GO3、TO2、TO1、AN3、AN2、AN1、GN1、CN1、GN2,与前面结构参数和结合能所得结论一致.

表2 (TPFC)Mn(Ⅲ)(L)的NPA电荷和韦伯键级Table 2 The NPA charge and Wiberg bond Index (WI) of (TPFC)Mn(Ⅲ)(L)
为理解(TPFC)Mn(Ⅲ)的Mn原子和碱基配位原子的轨道作用情况,对其进行二级微扰稳定化能(E(2))分析(表3),主要包括配位原子的孤对电子轨道(LP(N)或LP(O))与Mn原子的空电子轨道(LP*Mn)的作用. 由表3可知,ΣE(2)是α和β轨道的E(2)之和,但主要由β轨道的E(2)贡献. 配合物GO3中β轨道的E(2)由LP(L)对LP*(Mn,4s)和π*(O—Mn)的作用组成(π*(O—Mn)是碱基G的配位氧原子和(TPFC)Mn(Ⅲ)之间形成的O—Mn键π反键轨道),其它分子的α和β轨道的E(2)都由LP(L)对LP*(Mn,4s)的作用组成. ΣE(2)越大,配位原子和Mn原子之间的相互作用越强,即(TPFC)Mn(Ⅲ)和碱基之间的结合越强. ΣE(2)由186.98 kJ/mol(CO2)降至82.34 kJ/mol(GN2),ΣE(2)由大到小的顺序为:CO2、GO3、TO2、TO1、AN3、AN2、AN1、GN1、CN1、GN2,与结构参数、结合能以及WIMn—L数据分析的结论一致.
综上所述,DNA的4个碱基都可以单独与(TPFC)Mn(Ⅲ)进行轴向配位,(TPFC)Mn(Ⅲ) 和碱基之间的结合由强到弱为:CO2、GO3、TO2、TO1、AN3、AN2、AN1、GN1、CN1、GN2,每个碱基的配位能力都会因自身独特的结构而不同. 当以同一碱基的不同原子作为配位原子时,碱基上的O原子和Mn原子的结合能力最强;对不同的碱基而言,当以O原子配位时,(TPFC)Mn(Ⅲ)与C的结合最强,与T的结合最弱;当以N原子配位时,(TPFC)Mn(Ⅲ)与A的结合最强.

表3 (TPFC)Mn(Ⅲ)(L)的二级微扰稳定化能
2.2 (TPFC)Mn(Ⅲ)和碱基对的轴向配位性质
在(TPFC)Mn(Ⅲ)与DNA碱基的轴向配位性质理论研究的基础上,对(TPFC)Mn(Ⅲ)与A=T、C≡G碱基对的轴向配位性质进行理论研究,分子模型见图3,几何优化结构见图4,依次命名为AT1、AT2、AT3、AT4、AT5、AT6、GC1、GC2、GC3、GC4、GC5和GC6,AT1~AT6是A=T与(TPFC)Mn(Ⅲ)的配合物,GC1~GC6是C≡G与(TPFC)Mn(Ⅲ)的配合物. 在AT1~AT4及GC6中,碱基对在(TPFC)Mn(Ⅲ)平面的上方,近似于平行,表现为外部结合模式,AT1和AT2的2个碱基发生了扭曲,不再处于同一个平面上. 在AT5、AT6、GC1~GC5中,(TPFC)Mn(Ⅲ)插入碱基对之间,A、T(或G、C)分布在(TPFC)Mn(Ⅲ)平面的两侧,碱基对之间的氢键被破坏.

图3 (TPFC)Mn(Ⅲ)(A=T)和(TPFC)Mn(Ⅲ)(C≡G)的分子结构

图4 (TPFC)Mn(Ⅲ)与DNA碱基对的优化几何结构
(TPFC)Mn(Ⅲ)(A=T)配合物的部分结构参数和结合能ΔE见表4. 当(TPFC)Mn(Ⅲ)与A=T以外部结合模式形成配合物(AT1~AT4)时,由配位键键长dMn—O或dMn—N可知,AT1~AT3均形成有效配位,其配位键长分别为0.238 7、0.248 5、0.232 1 nm,AT4未形成有效配位. AT1~AT3的结合能ΔE分别为-3.65、-12.72、-16.64 kJ/mol,且在AT2和AT3中(TPFC)Mn(Ⅲ)均与A配位,说明在(TPFC)Mn(Ⅲ)以外部结合模式与A=T结合时,A的配位能力强于T.
当以插入结合模式形成配合物AT5、AT6时,均形成有效配位,A和T分别以N原子和O原子作为配位原子. 在AT5中,配位键长dMn—O和dMn—N分别为0.261 3、0.253 2 nm,在AT6中,dMn—O和dMn—N分别为0.264 1、0.2564 nm. 通过比较,dMn—O 表4 (TPFC)Mn(Ⅲ)(A=T)的部分几何结构参数和结合能Table 4 The selected geometrical parameters and binding energy of (TPFC)Mn(Ⅲ)(A=T) (TPFC)Mn(Ⅲ)(C≡G)配合物的部分结构参数和结合能ΔE见表5,当(TPFC)Mn(Ⅲ)与C≡G以插入结合模式形成配合物(GC1至GC5)时,C和G均分布在(TPFC)Mn(Ⅲ)的两侧,但不能同时与(TPFC)Mn(Ⅲ)配位;在GC1和GC2中均为(TPFC)Mn(Ⅲ)直接和G的配位,配位键长分别为0.220 4、0.220 2 nm;在GC3、GC4和GC5中均为(TPFC)Mn(Ⅲ)直接与C的配位,配位键长分别为0.243 9、0.243 4、0.221 1 nm,且(TPFC)Mn(Ⅲ)与G的|ΔE|均大于与C的|ΔE|,由此可知在插入结合方式中G的配位能力强于C. 而以外部结合时,在AT6中C≡G和(TPFC)Mn(Ⅲ)不能有效配位,且结构不稳定,因此(TPFC)Mn(Ⅲ)和C≡G的作用以插入结合方式为主. 表5 (TPFC)Mn(Ⅲ)(C≡G)的部分几何结构参数和结合能Table 5 The selected geometrical parameters and binding energy of (TPFC)Mn(Ⅲ)(C≡G) 在(TPFC)Mn(Ⅲ)与碱基对配位形成的配合物中,选取稳定性较好的配合物作为代表进行NPA分析(表6),即对AT1、AT3、AT5、GC1、GC4和GC5进行分析讨论. 在A=T碱基对中,外部结合的AT1和AT3分别是碱基T和A与(TPFC)Mn(Ⅲ)配位,AT3的ΔQL大于AT1,且AT3的ΔQA(0.156 e)大于AT1的ΔQT(0.120 e),说明A的配位能力强于T;在插入结合中,AT5是A和T同时与(TPFC)Mn(Ⅲ)配位,ΔQA(0.130 e)大于ΔQT(0.111 e),可知在A=T碱基对中,A的配位能力强于T. 在C≡G碱基对中,对于GC1、GC4、GC5分别是G、C、C与(TPFC)Mn(Ⅲ)配位,各配位碱基电荷转移量分别为0.192 e、0.140 e、0.156 e,可知在C≡G碱基对中,G的配位能力强于C. 表6 (TPFC)Mn(Ⅲ)(A=T)和(TPFC)Mn(Ⅲ)(C≡G)的NPA电荷Table 6 The NPA charge of (TPFC)Mn(Ⅲ)(A=T) and (TPFC)Mn(Ⅲ)(C≡G) QA、QT是形成A=T碱基对后A与T的电荷,QG、QC是形成C≡G碱基对后G与C的电荷,在配位之前,QA、QT、QG和QC分别为0.019 e、-0.019 e、-0.055 e和0.055 e. 采用DFT/B3LYP 的方法对(TPFC)Mn(Ⅲ)与DNA的4种碱基以及碱基对的轴向配位性质进行理论研究. 计算结果表明:以相同碱基的不同原子作为配位原子时,碱基上的O原子与(TPFC)Mn(Ⅲ)配位能力最强;对不同的碱基而言,当配位原子为O原子时,(TPFC)Mn(Ⅲ)与C的结合最强,与T的结合最弱;当配位原子为N原子时,(TPFC)Mn(Ⅲ)与A的结合最强. 对比研究与A=T和C≡G碱基对轴向配位的结果表明:(TPFC)Mn(Ⅲ)主要以插入方式与A=T和C≡G结合;对于A=T碱基对,A的配位能力强于T;对于C≡G碱基对,G的配位能力强于C.


3 结论
猜你喜欢
科学之谜(2021年2期)2021-04-25
山东科技大学学报(自然科学版)(2021年2期)2021-04-10
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
科学导报(2020年54期)2020-09-09
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
当代陕西(2019年6期)2019-04-17
分析化学(2018年2期)2018-03-02
新高考·高一物理(2016年7期)2017-01-23
新高考·高一物理(2015年6期)2015-09-28
