
CO2 高值化利用新途径:铁基催化剂CO2加氢制烯烃研究进展
2021-03-08 01:21张超张玉龙朱明辉孟博涂维峰韩一帆
化工进展 2021年2期
张超,张玉龙,朱明辉,孟博,涂维峰,韩一帆,
(1 化学工程联合国家重点实验室,华东理工大学,上海200237;2 先进功能材料制造教育部工程中心,郑州大学,河南郑州450001)
煤、石油和天然气等化石能源的开发与利用为人类的繁荣发展做出了重大贡献,但也导致大气中CO2浓度逐年攀升。根据美国夏威夷莫纳罗亚天文台(Mauna Loa Observatory) 观测的数据,截至2018 年7 月,大气中CO2浓度已达到410.8μL/L,该数值是工业革命前(18 世纪60 年代)278μL/L的1.48 倍[1]。CO2通常被认为是温室气体,会导致全球变暖和海洋酸化等问题[2-4]。根据政府间气候变化小组此前发表的评估报告,到21 世纪末大气中CO2的浓度或将达到560μL/L,届时全球平均温度将较工业革命时期上升1.5~4℃[5];根据英国科学家杰伦特-塔林的研究结果,从工业革命时期至今,全球海水表层的pH已经降低了约0.3。如果海洋酸化问题持续加重,将导致大量海洋生物濒临灭绝[6-7]。由此可见,CO2的排放问题不容忽视。
我国是全球第一大CO2排放国。根据BP 公司统计数据,2017年我国CO2总排放量达92.33亿吨,其中钢铁、水泥、化工三大行业CO2排放量占全国碳排放总量的50%左右[8]。由于我国是一个“富煤、贫油、少气”的国家,约70%的能源消耗直接依赖于煤炭的燃烧与加工[9]。此外,我国对于石油和天然气的需求亦是逐年递增。根据中国石油新闻中心的报道,2018 年国内原油对外依存度达70.9%,天然气达43.4%[10],考虑到工业周期性回暖叠加消费升级,我国对化石能源的需求依旧十分强劲[11]。由此可见,倘若我国对化石能源消耗不加以控制,CO2排放量将继续增加。
为了缓解CO2排放所带来的环境问题,国务院此前发布《国家中长期科学和技术发展规划纲要》(2006—2020)中明确提出了突破清洁能源技术,优化能源结构的发展目标。2016 年4 月,中国等175个国家签署了应对全球气候变化行动的《巴黎协定》。中国作为发展中国家已承诺到2030年单位GDP的CO2排放量要比2005年下降60%~65%,非化石能源在总的能源消费中所占比例要提升到20%左右。
控制CO2排放目前主要集中在新能源的开发以及可再生能源的利用,但值得注意的是,虽然CO2是一种温室气体,但是其本身也是一种重要的资源。根据中国碳交易网数据,我国八省市2019 年碳市场配额累计交易6.96千万吨二氧化碳当量,累计成交总额是约15.62亿元人民币,比2018年同比增加11%、24%[12]。随着国内各个省市对排放碳资源的合理化利用逐步普及,国内排放碳资源化利用市场产值甚至可超千亿元。
2012年,国际战略委员会将此前提出控制CO2排放的碳的捕获与封存(carbon capture and storage,CCS)战略修改为碳的捕获与利用(carbon capture and utilization,CCU)战略。CCU 是将化石能源燃烧释放的CO2捕获后进行资源化利用,该过程可以产生经济效益,更具有现实操作性[13]。
当前,CO2的资源化利用主要包括物理和化学方法[14]。物理方法利用其性质而不改变其化学结构,如气态CO2可用作食品添加剂或灭火气氛等;固态CO2(干冰)广泛用于防腐领域。化学方法则是将CO2通过工业反应转变为高附加值化学品的过程,是实现CO2循环利用的最佳途径之一,对提高碳的利用率及化石资源的高效洁净利用具有重要战略意义。
1 CO2加氢反应
CO2资源化利用制取的高附加值化学品主要包括甲醇、甲酸、二甲醚、烯烃、高碳醇、液体燃料等(图1)[15],而这些高附加值化学品的生产过程中皆需借助H2作为原料。目前,化工领域氢气的来源除去煤化工用煤和水生产合成气(C+H2O)的方法外,还包括石油气炼化裂化、氨分解、氯碱工业以及甲醇重整及炼焦炉废气(焦炉气中氢气成分占比55%~60%)等[16]。由此可见,中国当前化工工业具有强大和广泛的制氢基础。上述产氢过程中富余的氢气可作为部分CO2加氢资源化利用的H2来源。根据莫尼塔研究报道,2015 年国内副产氢气的商用剩余量约40 万吨/年。此外,中国有近200万吨/年的潜在专业制氢富余产能可做后续氢源供应[17]。在不考虑物流运输带来额外费用的前提下,国内每年总计约有240万吨的氢气供应无需新增资本投入便可加以利用(图2)。因此,在当前中国氢能经济发展的初期阶段,可在副产氢充足的地区进行CO2加氢制备高附加值化学品。
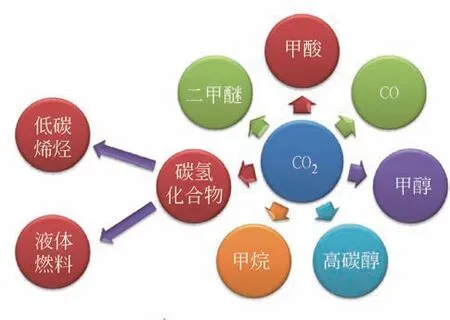
图1 CO2制取高附加值化学品
低碳烯烃作为重要的化工原料,CO2加氢制取低碳烯烃具有重要意义。乙烯工业是一个国家石化产业发展的最重要的标志,并且乙烯是全球需求量最大的烯烃之一,其下游衍生物应用领域十分广泛,涉及汽车、建筑、物流、农业、日用容器、服装鞋子、电力线缆、家电和医疗等,与国民经济各领域的增长紧密相关。根据国家统计局数据,乙烯表观消费量已从2010 年的1497.1 万吨增长至2017年的2036.9万吨,年均复合增长率分别为3.64%和4.50%;在进口方面,2010 年我国乙烯进口量81.5万吨,2017年乙烯进口量215.7万吨,进口规模总体扩大。另一方面,丙烯需求情况同样旺盛。2017年,国内丙烯表观消费量达到3140 万吨,近十年年均复合增长率达到11%以上[18]。随着国内产业升级及人民生活水平的日益提高,乙烯、丙烯产业高景气状况有望长期维持。
我国乙烯、丙烯的生产过程中面临的主要矛盾是自给率水平较低、常年呈现供不应求的局面。2018 年乙烯单体净进口量258 万吨,下游聚乙烯、乙二醇等产品净进口折算当量2380 万吨。2018 年乙烯表观消费量2630 万吨,当量消费量达到5010万吨,而总产能为2550 万吨/年,当量缺口超过2000 万吨/年,当量自给率不足50%[19]。根据石油和化学工业规划院此前发布的《我国烯烃产业链现状及“十三五”展望》所述,2020 年我国乙烯产量有望达到3040 万吨,但需求量也将提升至4800万吨[18],供需平衡差将继续拉大。丙烯方面,尽管国内丙烯产能产量近些年受益于丙烷脱氢、石油炼化、煤化工等项目大幅扩产,需求端对外依存度大幅下降。但截至目前,国内丙烯市场仍存在10%~20%的供给缺口。由此可见,开发不同路径的烯烃生产技术及产能扩充具有重要的现实意义。
生产工艺方面(图3),当前全球乙烯的生产工艺主要通过石脑油裂解(43%)和乙烷脱氢(36%),而丙烯生产则主要来源于原油催化裂化(35%)和石脑油的蒸汽裂解(32%)。然而,生产烯烃过程中占比极高的石脑油蒸汽裂解工艺过程是石油化工中的高能耗装置,反应温度在800℃以上,耗水量巨大,而且该工艺每生产1t 低碳烯烃需要消耗大约3t 的石脑油原料,折算下来每吨烯烃产品需要耗费约10t的原油,而且该过程完全依赖不可再生的石油资源。随着国内石油资源日益减少,对外依存度逐渐加大,利用CO2加氢制取低碳烯烃不但可以实现CO2的资源化利用,同时生产出的低碳烯烃还可以缓解我国石油化工资源不足所带来的压力。
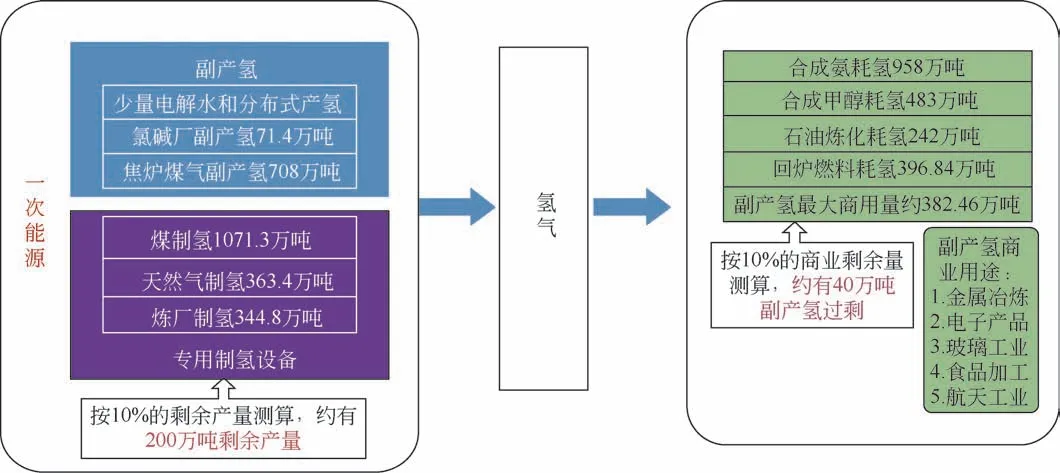
图2 国内制氢领域氢气富余情况[16]
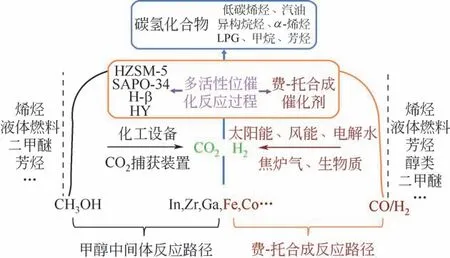
图3 CO2加氢制取烃类产品的途径[32](甲醇中间体路径和CO中间体路径)
但是,CO2加氢制烯烃反应目前尚未实现工业化。当前关于CO2制取烃类化学品的研究主要集中在三个领域,分别是光催化领域[20,21]、电催化领域[22-23]以及热催化领域[24-25]。由于电催化以及光催化法转化CO2制备烃类化学品的方法存在生产成本高以及工艺不成熟等问题,在可预见的时间内难以实现大规模工业化生产。与上述两种方法不同的是,热催化转化法是当前工业催化领域应用最为广泛的方法,具有工业化前景,因此研发高活性和高低碳烯烃选择性催化剂成为了推动CO2加氢工业化生产的关键。
2 催化反应分析
2.1 CO2加氢制烯烃的合成路径
在20 世纪20 年代末期,Hans Fischer 和Franz Tropsch两位科学家首次发表了关于通过使用CO和H2合成烃类化学品的报道[26]。此后,Fischer-Tropsch合成(费-托合成)便被认为是石油来源生产燃料和化学品的替代途径[27-28]。由于CO2催化加氢制备烃类(包括低碳烯烃和长链烃)产品本身就相当于费-托合成反应的延伸,即用CO2代替CO进行催化合成反应,因而该合成方法也逐步吸引了来自学术界和工业界的关注。
目前,关于CO2加氢制取烃类化学品采取的合成路径主要分为两类:第一种路径是甲醇中间体路径[29,30],即先将CO2转化为甲醇中间产物,随后通过甲醇耦合脱水制取烃类化学品;第二种路径是CO 中间体路径[31-32],即将CO2转化为CO,随后进行CO 加氢反应以合成烃类化学品。第一种路径本质上相当于间接法合成路径,该路径催化材料的选择主要集中在利于生产甲醇的铟基[33-34]、锆基[35-36]、镓基[37-38]催化剂与利于甲醇脱水聚合的HZSM-5[39-40]、SAPO-34[41-42]等分子筛材料的联用;第二种路径则相当于是直接法合成路径,所采用的催化剂同费-托合成催化剂类似,主要集中在Fe基[43-44]以及Co基[45-46]催化材料上。
2.2 热力学分析
CO2加氢制取低碳烯烃的反应过程主要考虑烯烃产物进行热力学分析。下文讨论CO2加氢制取烯烃过程中反应温度、反应压力以及CO2与H2的摩尔比对平衡组成的影响。
2.2.1 温度对CO2加氢反应热力学的影响
图4 显示了在无烷烃产物体系中CO2加氢反应过程中反应温度(反应温度区间为100~900℃)和反应平衡转化率以及烯烃选择性之间的关系[49-50]。从图4 中可以看出,当反应温度位于100~450℃时,CO2的转化率逐渐下降;而当反应温度高于450℃时,CO2转化率逐步上升。产物选择性方面,当反应温度在300℃以下时,逆水气变换反应产物CO 的选择性非常低(<5%),但是烯烃的选择性非常高(>90%)。随着反应温度上升至300℃以上,逆水气变换反应活性增加;当反应温度达到600℃时,CO 的选择性甚至可以接近100%,而与之相对的烯烃选择性则随着反应温度的进一步上升而不断下降。
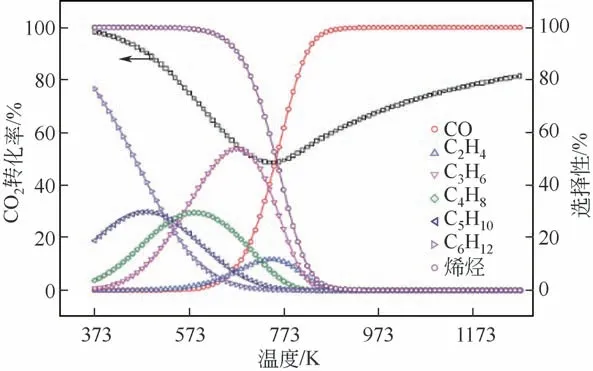
图4 不同温度下CO2转化率及产物选择性的关系[47]

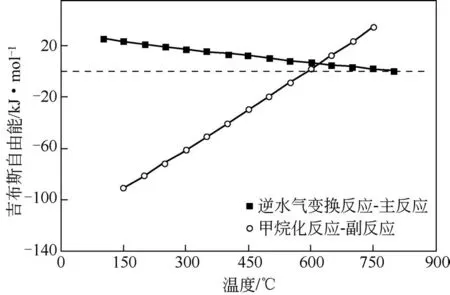
图5 逆水气变换反应和甲烷化反应吉布斯自由能与温度的关系[48]
反应转化率方面,由于CO2加氢反应整体为放热过程,在较低温度条件下,体系中合成烯烃的反应路径占据主导,因此在低温段随着反应温度的升高,CO2的平衡转化率降低。而在高温条件下,由于逆水气变换反应在体系中占据主导,该反应本身为吸热过程,因此在高温段随着反应温度的升高,CO2的平衡转化率升高。
2.2.2 压力对CO2加氢反应热力学的影响
图6 展示了不同反应温度条件下(250℃、300℃、350℃和400℃)CO2加氢平衡转化率与反应压力(0.1~5MPa)之间的关系。从图6 中可以看出,当反应压力从0.1MPa 上升至2.0MPa 时,CO2的平衡转化率急速上升;当反应压力上升至2.0MPa 以上时,平衡转化率上升逐渐趋于平缓。
烯烃整体选择性的变化规律与反应平衡转化率随压力的变化规律一致,但涉及具体烯烃产物,选择性与压力的变化关系则略有不同:乙烯的选择性在不同反应温度条件下(250~400℃)均随反应压力的上升而提高;而丙烯选择性随压力的变化规律则与乙烯不同,其在250~300℃温度段[图6(a)、(b)],会随着压力的上升而下降,当温度上升至350~400℃时[图6(c)、(d)],丙烯的选择性则会先上升后下降。另外,通过对压力与烯烃选择性的关系分析比较可以发现,当反应压力在1.5~2.5MPa 之间时,反应最有利于C2~C4烯烃生成。
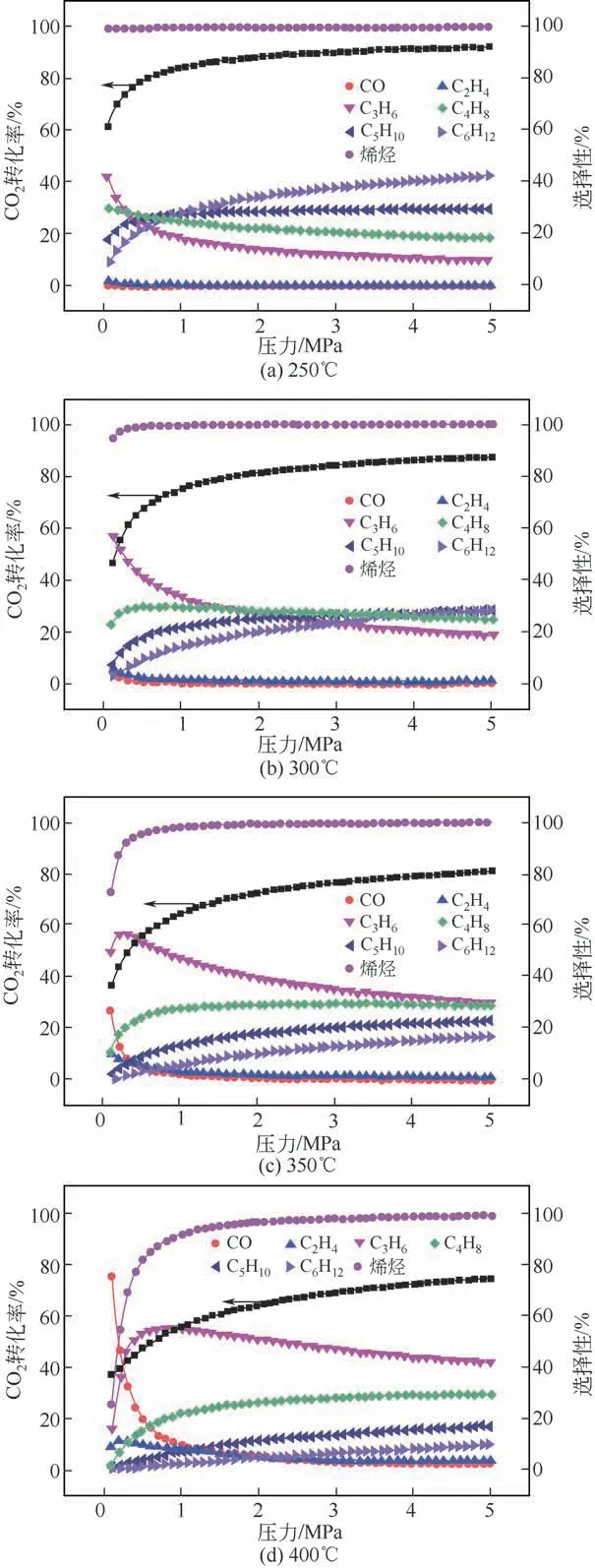
图6 热力学平衡条件下,反应压力与CO2加氢转化率和产物选择性随温度变化的规律[24]
2.2.3 CO2和H2摩尔比对CO2加氢反应热力学的影响
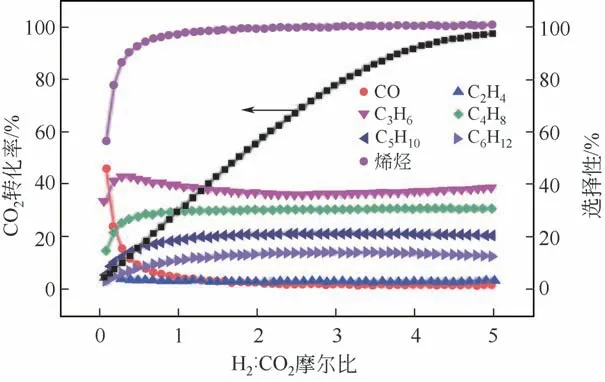
图7 350℃下,H2与CO2摩尔比对平衡条件下反应转化率及产物选择性的影响[49]
图7反映了CO2和H2摩尔比对催化剂活性及产物选择性的影响[49]。随着H2/CO2摩尔比的增加,CO2的转化率始终呈现上升趋势;同时随着H2比例的逐步升高,产物中CO 和烯烃的选择性呈相反趋势发展,即烯烃选择性逐步升高,CO选择性逐步下降。另一方面,烯烃与CO选择性变化主要集中在H2/CO2摩尔比在0~1之间;当比率大于1时,CO及烯烃的整体选择性趋于稳定。通过图7可以判断,制取低碳烯烃应采用H2/CO2摩尔比大于1的原料气进行反应。
Arena 等[25]也对CO2加氢反应制备低碳烯烃进行了热力学研究,重点分析了反应产物在C2~C4的烯烃产物。通过热力学计算,总结出最适用的CO2加氢制取低碳烯烃的反应条件为:温度307~357℃之间,反应压力1.5~3.0MPa 以及H2/CO 摩尔比为3。该反应条件与前文对CO2加氢制取烃类产品的条件筛选结论类似。此外,他们还计算出在上述反应条件下,CO2的理论最高转化率为70%左右。
2.3 CO2加氢产物分布理论模型
由于CO2加氢反应可以被认为是修饰的费-托合成反应,因而反应过程中同样存在碳-碳偶联链增长与碳链加氢脱附之间的平衡关系,这意味着在CO2加氢制取烃类产品的反应中,具体的产物分布同费-托合成反应一样宽泛。为了对费-托合成以及CO2加氢反应过程产物分布进行描述,研究人员于1951 年提出了Anderson-Schulz-Flory(ASF)分布理论模型[51],该模型假定当反应达到平衡状态时,相同碳原子数的各种不同化合物碳链增长速率和终止速率相同的情况下,不同碳数的产物分布的理论值可以通过式(1)进行计算,基于该公式计算的具体产物分布情况如图8(a)所示。
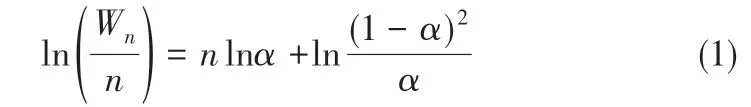
式中,Wn为产物碳基质量分数;n为产物含碳个数;α为产物的链增长因子。
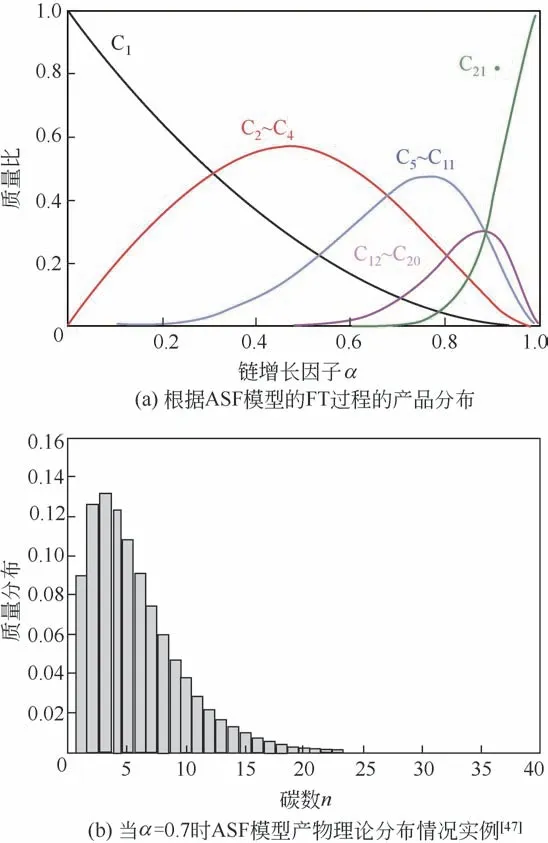
图8 费-托合成以及CO2加氢反应过程产物分布
图8(a)直接反映了费-托反应及CO2加氢反应存在的本质问题,即由于产物分布范围广,反应本身不利于获得高选择性的烃类目标产物。例如,当链增长因子(α)为0.7时,C2~C4低碳烃类产品的理论选择性上限仅为38%,液体燃料(C5+)的理论选择性上限为53%[图8(b)]。
由此可见,如何打破ASF分布成为提高反应目标产物选择性的关键。此外,CO2加氢同CO 加氢过程不同,由于CO2分子本身是直线型结构,化学性质较CO更为稳定,CO2加氢反应过程中的碳-碳偶联活性相对较低,不利于反应生成所需的烃类目标产物(C2+烃类)。因此,大多数CO2加氢反应产生的主要烃类产物为甲烷。为了解决上述两大问题,研究人员对各种催化剂进行了改性。
2.4 铁基催化剂的设计与开发
考虑到CO2加氢反应过程中烃类产品主要通过碳链的偶联增长形成,催化剂的选择可以借鉴CO加氢制取烃类化学品常用的催化剂开展研究。由于Fe基催化剂和Co基催化剂是传统的费-托合成催化剂,因而在CO2加氢过程中同样可以被广泛使用。但是,与CO 加氢催化过程不同的是,CO2加氢反应过程并不是直接发生碳-碳偶联的。反应过程首先要经历逆水气变换过程生成CO的中间产物[反应式(2)],然后再通过费-托合成生成碳氢化合物[反应式(3)]。同Co 基催化剂相比,Fe 基催化剂由于具有更强的逆水气变换活性[52],因而其在CO2加氢制烃类化学品的反应过程中具有相对更高的催化活性。因此,Fe基催化剂被认为是更具有实现CO2加氢制烯烃工业化前景的催化材料。
虽然Fe催化剂对CO2加氢具有良好的性能,但单组分Fe 催化剂的性能上还远远不能达到实际应用的要求。为了提高Fe 催化剂的反应活性和烃类产品选择性,目前主要研究重点包括催化剂载体、助催化剂掺杂改性以及反应活性及反应机理研究。下文将着重讨论当前Fe基催化CO2加氢反应过程的研究现状,分析助催化剂、载体、催化剂结构等因素对反应活性及产物选择性的影响。
2.4.1 助催化剂对Fe基催化剂的影响
(1)碱金属助催化剂 一般情况下,CO2的吸附发生在催化剂的碱性位点上,因此Na、K、Cs等金属元素作为有望促进CO2吸附和增强反应活性的助催化剂材料被广泛使用[53-56]。在提高低碳烯烃选择性方面,碱金属的加入通常是必不可少的,由于碱金属可促进CO2的吸附,而H2的吸附能力则间接被抑制[57]。因而,碱金属的加入在提升催化活性的同时,也提高了低碳烯烃选择性。当前关于碱金属助催化剂的报道中,K和Na被认为是提高活性及烯烃选择性最有效的元素。
Choi 等[55]的研究显示,掺杂了助催化剂K 的Fe-K催化剂在进行CO2加氢反应过程中,CO2更倾向于吸附在K 原子位点上,而H2则倾向于吸附在Fe 原子位点上。由于助催化剂K 本身更为偏好吸附CO2,而对H2的吸附则相对排斥,增加了反应过程中碳-碳偶联的概率,而催化剂表面C—C 键的形成和H2浓度的降低有利于生成烯烃。
国内外关于碱金属助催化剂用于CO2制低碳烯烃已经取得了一些进展。此前,厦门大学王野教授等[53]还发现,通过助催化剂K改性的Fe催化剂中添加适量的B元素可以在不改变催化剂活性的情况下进一步提高低碳烯烃选择性;宁夏大学的赵天生教授团队[58]通过采用微波辅助水热法制备的含有K助催化剂的Fe 基催化剂C2~C4烯烃选择性达到了53.58%。此外,大连化学物理研究所的孙剑团队[59]还发现,加入碱金属Na 的Fe3O4催化剂在进行CO2加氢实验中同样具有较高的反应活性和烃类选择性,其C2~C4烯烃和C5+烃类产物选择性分别可达46.6%和30.1%。
为了考察碱金属助催化剂对催化剂结构与反应性能的影响,近期,Guo 等[60]通过对含有碱金属元素的生物助催化剂修饰的Fe 基催化剂在反应前后进行了穆斯堡尔谱(Mössbauer)和X 射线光电子能谱(XPS)表征,发现催化剂中的K元素会随着反应在催化剂表面发生迁移,而K 的迁移会促进催化剂表面铁元素的碳化,同时K 的碱金属特性还会降低反应过程中催化剂对氢气的吸附,从而使最终产物中的烯烃选择性得到提高,如图9。Kangvansura 等[61]使用in situ XANES 谱 研究了K/Fe/NCNT 和Mn/Fe/NCNT 在还原过程中,从323K 到653K 保持300min 过程中的结构变化,图10 显示K/Fe/NCNT 在还原300min 后,Fe、FeO、Fe2O3和Fe3O4相均可检测到。300min后在Mn/Fe/NCNT中只检测到FeO 和Fe3O4。结合性能表明添加K 助催化剂的Fe/NCNT 可提供碳化铁形成的活性铁,从而成为CO2加氢制烯烃和醇的活性中心。由此可见,碱金属助催化剂的改性是提高催化活性和烯烃产物选择性的有效手段。
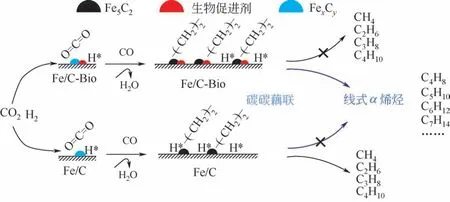
图9 Fe/C和Fe/C-Bio催化剂用于CO2加氢制取线性α-烯烃的反应示意图[56]
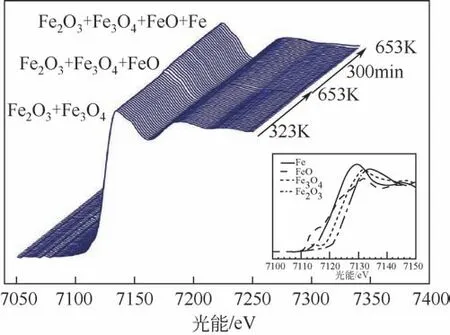
图10 K/Fe/NCNT和Mn/Fe/NCNT在还原过程中从323K到653K保持300min过程中结构变化的in situ XANES谱[61](插图为Fe、FeO、Fe3O4、Fe2O3的标准谱图)
另一方面,尽管助催化剂的添加可以促进CO2的吸附进而提高反应活性,但是反应过程中形成的烯烃产物也会在反应过程中发生二次吸附,使得烯烃产物经历二次加氢反应转化为烷烃产品,最终降低了低碳烯烃的选择性。Lee等[62]在Fe基催化剂中添加助催化剂Ru 的研究中发现,Ru 的加入会使得反应过程中烯烃产物再吸附能力增强,进而加剧了产物的二次聚合或加氢。上述结果说明Ru 助催化剂的加入并不利于CO2加氢反应中低碳烯烃产物的生成,但是Lee 等认为Ru 助催化剂的加入可能有利于CO2加氢制取长链烃类化合物。关于Ru助催化剂的认识,Niemela 等[63]提出了相反的看法,他们认为Ru 助催化剂无论对CO2加氢催化反应活性和产物中的烯烃选择性的影响都微乎其微。
(2)过渡金属助催化剂 除了此前讨论的碱金属助催化剂,过渡金属如Mn、Zn、Cu等助催化剂由于具备促进Fe 基催化剂的还原以及增加催化剂表面碱度、颗粒分散性等优势,同样被广泛应用于CO2加氢反应过程[64-68]。
助催化剂Mn 通常被认为既是电子助催化剂又是结构助催化剂[67]。Dorner 等[15,65]认为Mn的加入一方面能够覆盖Fe 基催化剂的加氢位点,从而提高烯烃选择性。另外,反应过程中Mn原子可能还会沉积在链增长的位点上,抑制长链烃类产物的生成,提升低碳烯烃选择性。李永旺团队[69]的研究结果与前者类似,他们认为催化剂表面Mn原子的富集能够有效地降低反应过程中的链增长以及中间产物二次加氢的能力,提高低碳烯烃的选择性,如图11。
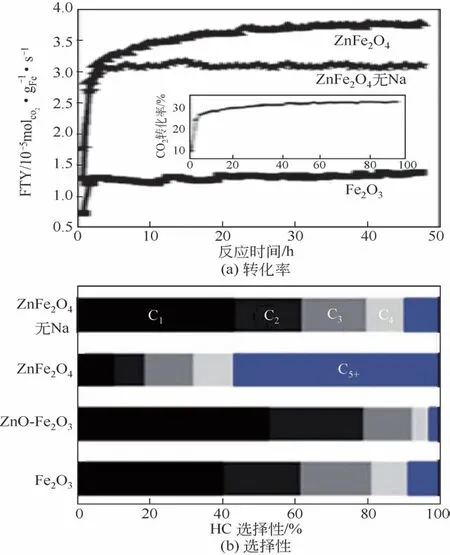
图11 Fe2O3、ZnFe2O4和不含Na的ZnFe2O4催化剂用于CO2加氢反应过程中转化率及选择性[69]
相对于Mn助催化剂,Zn助催化剂也可以促进Fe 催化剂表面的碱性,提高对CO2的吸附能力[70]。然而Zn 元素通常被认为是一种结构助催化剂,可以促进Fe 相的分散从而增加催化剂的比表面积,进而提升CO2加氢反应的活性。根据Zhang 等[58]报道,Zn 能够提高低碳烯烃的选择性,归因于Zn 与Fe 形成了ZnFe2O4固溶体结构,而该固溶体结构增强了Fe与Zn之间的相互作用,改变Fe元素周围的电子云密度,降低了反应过程中催化剂的加氢能力。Zhang 等[58]还发现Mn 和Cr 等助催化剂虽然也可以与Fe 形成固溶体,但是Zn 与Fe 形成的ZnFe2O4具有较高结构稳定性。关于助催化剂Zn的作用,在费-托合成过程的研究中,研究者们同样认为Zn 的作用并不仅仅是结构助催化剂,他们发现Zn 元素在改善催化剂分散性的同时,还作为电子助催化剂改善了催化剂表面的碱性,增强反应过程中低碳烯烃的选择性[71-73]。近期,Choi 等[74]制备了一种同时含有Zn 和Na 助催化剂的ZnFe2O4尖晶石结构的催化剂前体,该催化剂在经过活化后进行CO2加氢反应,可以生产出具有高烯烷比的液相烃类产品(图11)。
宋春山团队[75]制备了Fe-Cu双金属催化剂,相比单Fe 催化剂,由于形成Fe-Cu 合金,Fe、Cu 之间存在强烈的相互作用,可以提高CO2转化率并且抑制甲烷生成,有利于C2+烃的生成。Choi等[76]利用类似尖晶石结构的CuFeO2作为前体,得到了约65%的C5+选择性和2%~3%的甲烷选择性,他们归因于该前体的结构更有利于生成Fe5C2,其是生成C5+产物的活性相。
Ce 元素改性Fe 基催化剂用于CO2加氢反应同样有所提及[77-78]。由于CeO2具有良好的储氧能力,因而Ce 通常被认为是可以提高逆水气变换反应的优良组分。Williams等[64]通过对Ce煅烧温度以及颗粒尺寸进行控制,成功合成了同时具有逆水气变换活性和费-托合成反应活性的Fe-Ce双功能催化剂,该催化剂可以显著提高CO2加氢反应的转化率以及C2+烃类产物的选择性。
2.4.2 载体对Fe基催化剂的影响
除了前文所阐述的关于助催化剂的研究,载体的选择对于催化剂的开发同样十分重要。当前,对于载体的选择主要集中在氧化物载体上,包括Al2O3、TiO2、SiO2、ZrO2和CeO2等[79-81]。其中,载体Al2O3被证明可以提升CO2加氢反应活性[82],主要归因于其表面有着丰富的—OH基团,有利于对CO或CO2中间体的吸附。Xie 等[83]研究了载体孔径对CO2加氢制烯烃反应过程中催化性能的影响,发现Fe2O3颗粒的粒径会随着Al2O3载体的孔径增大而增大。此外,他们也通过调变载体孔径的方法探究了催化剂活性位点的分布与催化剂性能之间的关系,发现当Al2O3载体孔径在7~10nm 时,Fe2O3粒径分布会集中在5~8nm,而该颗粒尺寸下的Fe 基催化剂具有最优的CO2加氢催化活性及烯烃选择性。基于上述实验,Xie 等[83]还推测Fe 基催化剂颗粒大小的变化一定程度上会影响催化剂活性以及产物的分布情况。关于颗粒尺寸对催化剂性能的影响,荷兰乌特勒支大学的de Jong 教授团队[84]为此展开了研究,他们发现当Fe 基催化剂颗粒在7nm 以下时,粒径效应对催化剂活性及选择性的影响很大;而当Fe 基颗粒大于7nm 时,尺寸效应对催化剂性能影响相对较小(图12)。该结论同Xie 等对颗粒尺寸对催化性能影响的推测基本一致。
研究人员对Fe 基负载不同晶体结构的ZrO2载体及不同形貌的CeO2载体进行了CO2加氢研究。结果表明,单斜ZrO2晶型(m-ZrO2)对于CO2的吸附和活化效果要明显优于四面体ZrO2晶型(t-ZrO2)[85]。而CeO2方面,Torrente-Murciano 等[86]采用了3 种不同形貌的CeO2载体(颗粒形貌、棒状形貌和块状形貌)进行反应,结果显示棒状的CeO2载体制备的Fe 基负载型催化剂具有最高的烃类选择性,而块状的CeO2制备出的催化剂则具有最高的烯烷比。
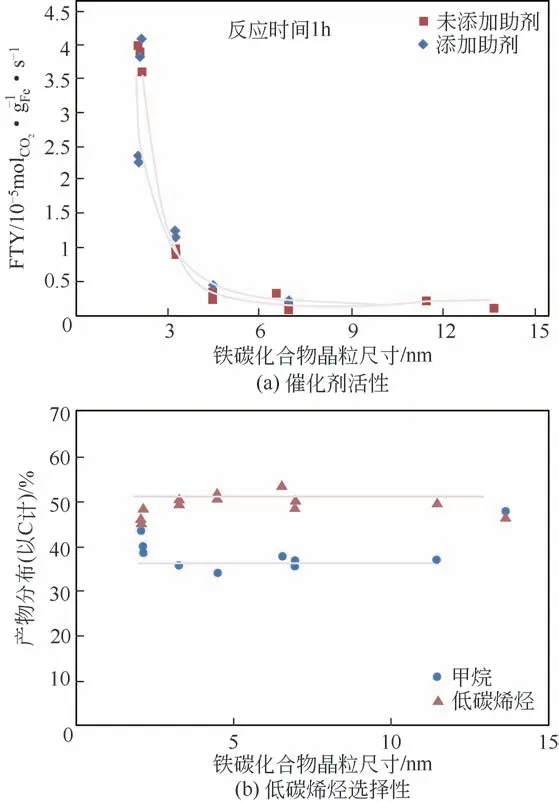
图12 不同碳化铁催化剂颗粒尺寸条件下的催化剂活性及低碳烯烃选择性[77]
研究表明,金属与载体之间的相互作用过强可能会抑制活化和反应过程中Fe相结构的碳化过程,一定程度上也会对催化剂的整体性能造成影响。因而,为了探究金属载体间的作用关系,除了此前陈述的一些常规的载体材料外,碳纳米管(CNTs)、核-壳碳材料和金属有机框架(MOFs)衍生物等新材料同样被广泛应用于CO2加氢反应的研究[87-89]。
研究表明,CNTs 上的纳米Fe颗粒在反应过程中具有良好的结构稳定性,这就意味着该类型催化剂在进行CO2加氢反应时不易失活[90]。另外,还有研究人员通过N 掺杂或者O 掺杂对CNTs 表面进行改性,从而改善纳米Fe 颗粒与载体间相互作用。研究结果表明,N 掺杂条件下的Fe/CNT 催化剂有利于提高催化活性,该结果可归因于纳米Fe 颗粒负载在N掺杂的CNTs比O掺杂的CNTs更易于被还原,从而形成活性相结构[91]。
Gupta等[92]对石墨化碳壳包覆Fe、Fe5C2和Fe3O4催化合成材料进行了研究,他们发现当石墨化碳相对含量从0.5 增加到1.5 时,催化剂活性会持续增加。然而,当纳米铁颗粒周围沉积的石墨化碳壳进一步增加时,催化剂活性开始下降,这可能是由于过量的碳层会阻碍反应物到达Fe活性位点的速率,因而催化剂活性出现了下降的趋势。此外,同Al2O3、SiO2等载体相比,氢气在石墨碳壳表面的迁移能相对较低,增强产物的加氢能力,最终导致烯烃的整体选择性下降(图13)。
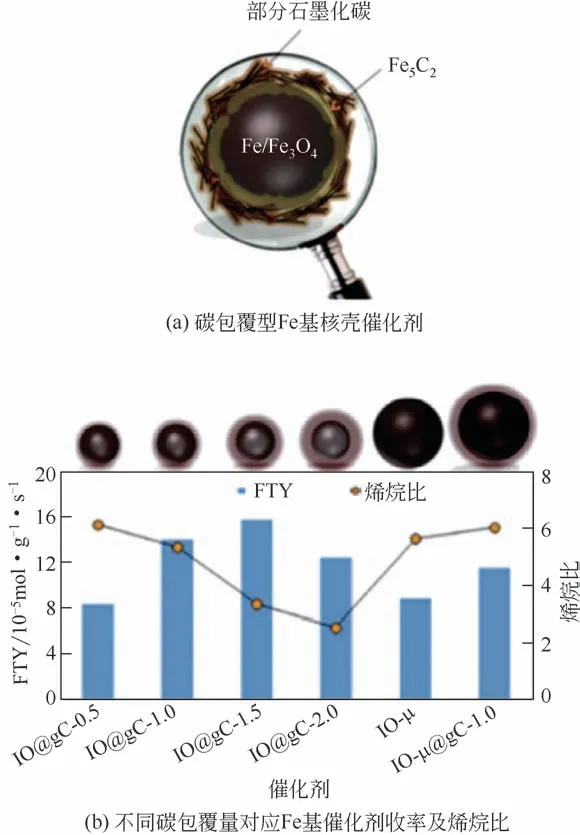
图13 不同碳包覆量Fe基催化剂的收率及烯烷比[92]
另外,关于MOFs 材料的研究同样被广泛报道。MOFs 材料由丰富的有机支板和排列有序的金属节点组成,该材料在退火后可形成含有金属修饰的碳纳米结构。MOFs 材料本身具有高比表面积、高孔隙率和择形催化等优点[93-94]。以Gascon团队开发的Fe-MIL-88B 催化剂为例,他们基于MOFs 材料衍生物为载体,采用热分解法制备了Fe 基多孔碳负载型纳米催化剂,该催化剂具有较高的催化活性,CO2单程转化率达到46%,反应产物中烯烷比可以达到5.5[95]。关于CO2加氢直接制取低碳烯烃Fe基催化剂中具有代表性的成果总结见表1。
通过上述讨论,当前对于CO2加氢制取低碳烯烃Fe 基催化剂助催化剂及载体的研究思路主要包括以下4种方式。
(1)添加碱金属助催化剂(如K、Na 等)增加催化剂的碱性位点,增加催化剂在反应过程中对CO2的吸附能力,从而提高催化剂整体的反应活性。同时,利用碱金属助催化剂对氢气吸附的抑制作用进一步增加烯烃选择性。
(2)添加过渡金属助催化剂(如Mn、Zn 等)改善Fe 基催化剂的分散性,增大反应过程中催化剂的比表面积,使得催化剂与CO2接触更加充分,从而提高催化剂活性。
(3)添加有利于逆水气变换活性的助催化剂(如Ce、Zn 等)。考虑到CO2加氢Fe 基催化反应路径通常被认为是先经过逆水气变换生产CO,而后是费-托合成制取烃类化合物,如果能够通过添加利于逆水气变换反应的助催化剂,同样可以起到促进反应活性的功效。
(4)添加有利于CO2吸附或利于Fe基材料分散的载体(如Al2O3、CNTs 等)或是优化载体与活性组分之间的相互作用,从而实现CO2加氢反应活性及低碳烯烃选择性的提高。
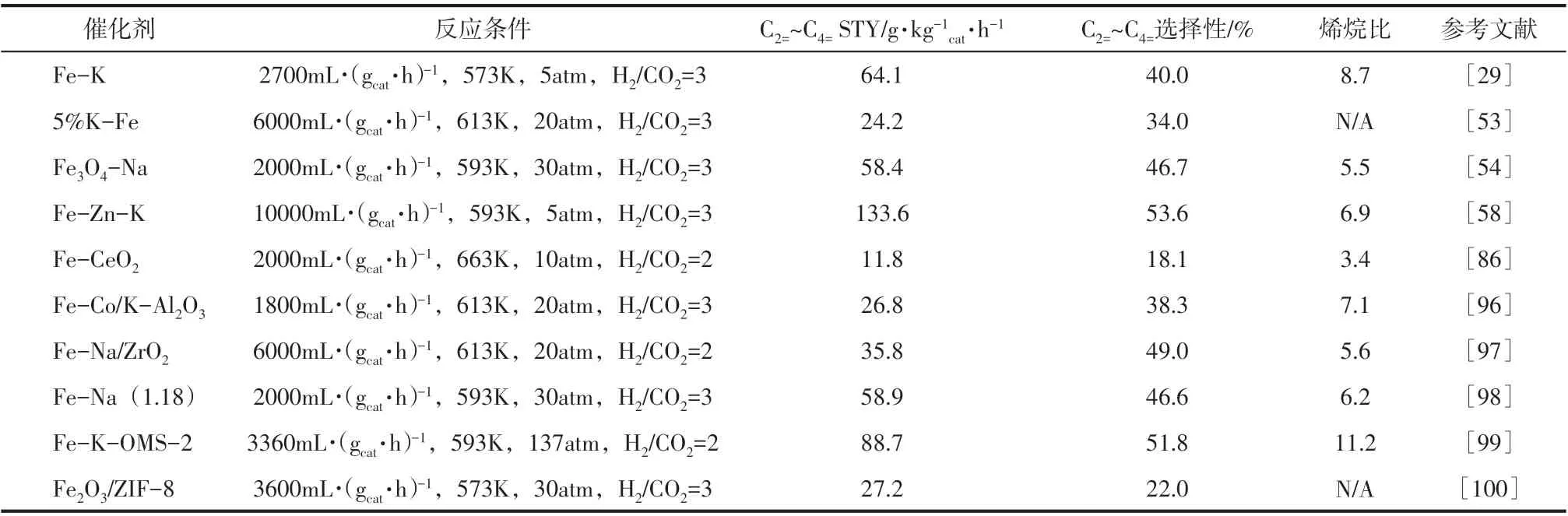
表1 CO2加氢制取低碳烯烃Fe基催化剂的工艺条件和反应性能
3 Fe基催化剂CO2加氢反应机理
3.1 CO2加氢反应机理
Lee 等[62]在2004 年提出CO2加氢Fe 基催化制取烃类化合物的反应机理。他们认为在反应过程中,CO2首先被催化剂中的Fe2+捕获活化,形成Fe3+(COO·)结构。随后H2中的H 自由基会分别与Fe3+(COO·)结构中的端基氧和羰基碳结合,形成羟基甲酸(HCOO·)或脱水生成Fe3+(CO·)。接下来H自由基会以与此前同样的方式继续与Fe3+(CO·)结构中的端基氧和羰基碳结合形成羟基甲醇(CH2O·)或进一步脱水生成Fe3+(CH2·)结构,而Fe3+(CH2·)结构中的CH2·便是碳链增长的引发物。另一方面,由于反应过程中长链烃类化合物是CO2加氢Fe基催化的主要产物,因而Lee等在提出的反应机理中特别强调了CO2插入Fe3+(CH2·)过程是影响反应链增长能力的主要因素(图14)。此外,他们还提出反应过程中烯烃与烷烃产物的调变机制可以归因于催化剂反应体系中H2的吸附量,即当H2吸附量相对富足,在可以满足羟基甲酸脱水形成的CH2·的同时,还可以进一步对产物进行加氢,则反应有利于烷烃产物的生成;但与之相对,当反应体系中H2的吸附量仅够满足吸附物种转化为CH2·结构时,反应将倾向于生成烯烃[101]。
另外,Meng 等[102]通过使用原位红外光谱研究了反应过程中CO2的吸附情况。他们所认为的烯烃生成机理同前文Lee 等对CO2加氢反应机理的认识相似。近期,以色列本古里安大学的Herskowitz 教授课题组[43,103]对CO2加氢Fe基催化剂的反应机理提出了与上述不同的观点,他们认为CO2加氢反应的第一步逆水气变换反应主要在Fe3O4上发生,而经过逆水气变换后生产的CO 则在FeCx结构上进一步与H2反应生产烃类化合物。具体的反应机理如图15 和图16 所示,CO2首先吸附在含有氧空穴位的Fe3O4结构上形成如图14所示的Fe(CO3)中间体,由于Fe(CO3)中间体中的Fe 与O 的相互作用强于C 与O 的相互作用,因而Fe(CO3)会断键脱除CO,随后剩余的O—Fe—O结构会与H2反应脱水再次生成含有氧空穴位的Fe—O结构,从而实现逆水气变换反应的循环过程。后续的烃类化合物的生成过程则发生在催化剂中的FeCx表面(图15),首先与H2反应生成烷烃产物,烷烃产物脱除形成含有碳空穴的FeCx结构。接着,此前逆水气变换反应过程中生成的CO 会占据FeCx结构中的碳空穴位点形成Fe—C—O 结构,该结构中的O 原子接下来会与H2反应脱水再次生成FeCx,从而实现CO 加氢生产烃类化合物的循环过程。
Riedel等[104]重点研究了反应过程中铁相结构的组分演变,通过将高分辨透射电子显微镜(HRTEM)等表征手段结合,他们将催化剂铁相结构反应过程中的结构演变历程分成了如图17 所示的5个阶段。其中第一阶段主要经历的是反应物吸附在催化剂表面并将Fe 相结构碳化的过程;第二和第三阶段,逆水气变换反应占主导,Fe 相结构碳化过程持续;第四阶段,Fe 相碳化过程进入稳定阶段,费-托合成反应占据主导;第五阶段,催化剂相组成结构趋于稳定,反应达到平稳状态。
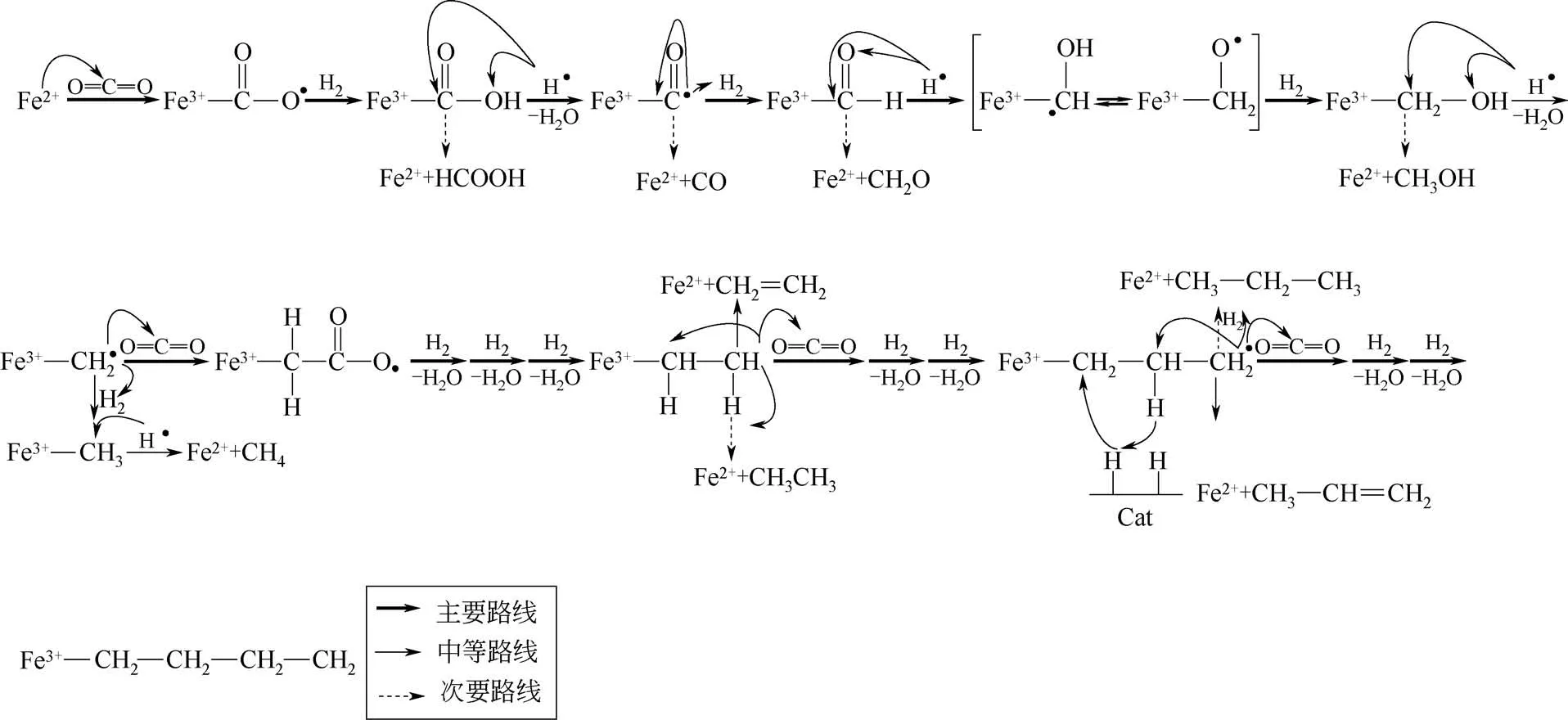
图14 CO2加氢铁基催化反应机理[94]
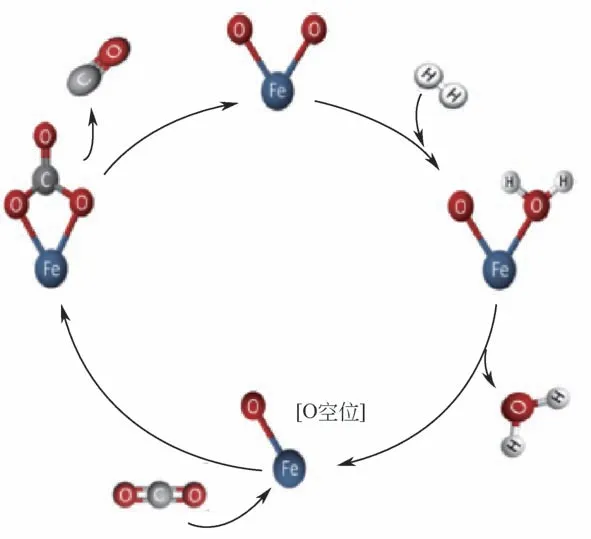
图15 Fe3O4表面催化CO2加氢逆水气变换反应机理[96]
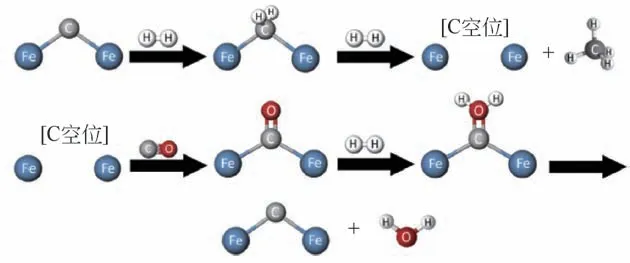
图16 碳化铁表面CO加氢制烃类化合物反应机理[96]
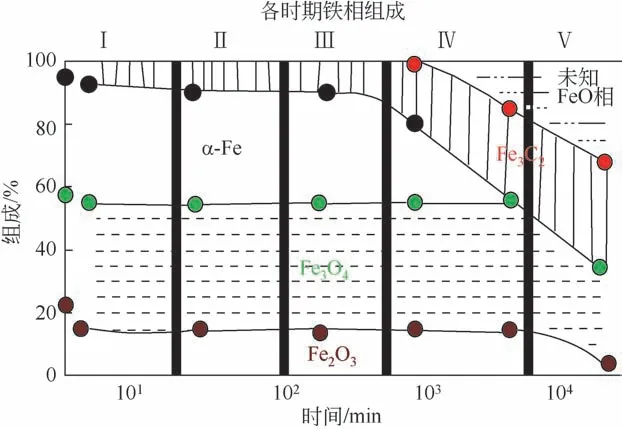
图17 CO2加氢制烃类化合物不同反应时期铁相组成结构[97]
在上述的结构演变过程中,催化剂中的α-Fe2O3结构会在反应初期逐步转变为α-Fe 和Fe3O4结构。随着反应的进行,催化剂中的Fe3O4相会先上升后减少。同时,一种新的无定形的铁氧化物相会随即生成,而该结构可能有利于逆水气变换反应的发生,而随后的费-托活性则始于碳化铁(FeCx)结构的生成而逐渐升高。
Zhu 等[105]通过in situ XRD 研究了不同粒径的Fe/ZrO2催化剂在预还原和CO2加氢反应过程中的动态结构变化,在纯氢还原过程中,温度由室温上升至400℃时Fe2O3相逐渐经FeO相转化为金属Fe,后切换至反应气氛围,金属Fe 转化为Fe5C2,小粒径的Fe2O3更难发生相转化。作者还用in situ DRIFT研究了CO2加氢中间体和CO 加氢反应过程中的(如图18),在CO2加氢时大粒径易生成HCOO*中间体,更有利于生成CH4,而HCO3*和CO3*在小粒径催化剂中更易生成,进而产生更多CO。在CO加氢过程中(如图19),红外中CH2、CH3中的C—H键证明烯烃和CHx*中间体在大粒径催化剂中利于形成,C—C耦联能力更强。
张玉龙等[106]通过Operando Raman 和Operando XRD 研究了不同Fe2O3前体(α-Fe2O3和γ-Fe2O3)在CO2加氢反应中的动态结构变化(如图20),发现α-Fe2O3经活化及反应过程倾向于生成χ-Fe5C2,有利于生成低碳烯烃;γ-Fe2O3经同样过程处理易生成θ-Fe3C,有利于生成C5+产物。
3.2 Fe基催化剂失活机理
前文重点讨论了催化剂的反应机理及反应初期阶段催化剂的结构演变规律。为了实现催化剂的放大和工业化进程,研究催化剂的失活原因同样十分重要。
Lee 等[107]通过XPS、HRTEM、Mössbauer 及程序升温氧化(TPO)等表征方法,重点考察了Fe-K/Al2O3催化剂在CO2加氢制取烃类化合物的失活机理。他们认为催化剂失活的原因与催化剂所在反应器的位置有关(图21)。其中,靠近反应器进口端的催化剂失活的原因主要归因于Fe5C2活性相转变为Fe3C结构,他们认为Fe3C结构是不利于CO2加氢反应的一种惰性FeCx结构。另外,通过TPO 和HRTEM 表征实验还发现,靠近反应器末端的失活后催化剂包覆有大量的积炭,同样是导致催化剂失活的原因之一[101]。
此外,还需要提及的是反应过程中发生的逆水气变换反应会使反应体系中生成大量的水,而水的生成同样会导致催化剂中的FeCx活性相结构氧化失活。因此很多研究人员在研究过程中试图将反应过程中生成的H2O 移除,以避免催化剂失活的同时提高催化剂的活性。这其中比较有代表性的例子同样出自于Herskowitz 教授课题组,他们自主设计了可将反应过程中生成的水移除的三段式反应器,成功地将CO2单程转化率从48%提升至89%[108]。
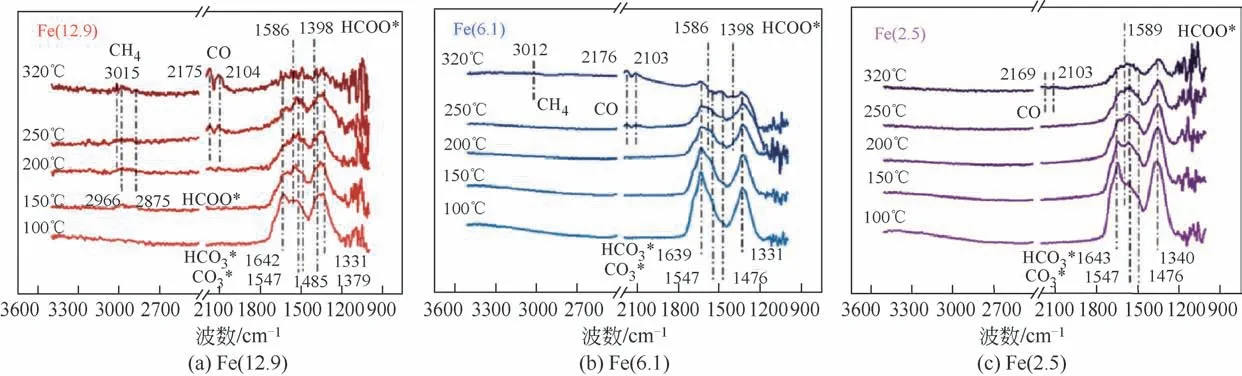
图18 Fe(12.9)、Fe(6.1)、Fe(2.5)在CO2加氢过程中DRIFT谱[105]
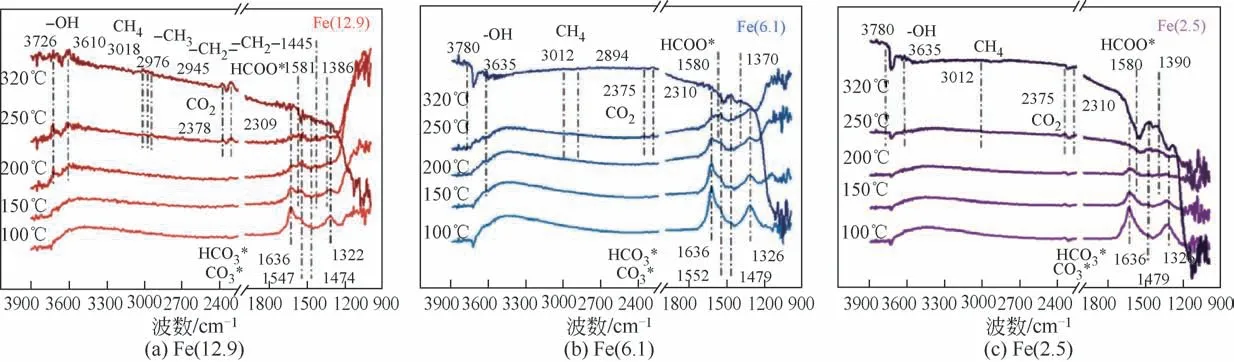
图19 Fe(12.9)、Fe(6.1)、Fe(2.5)在CO加氢过程中DRIFT谱[105]
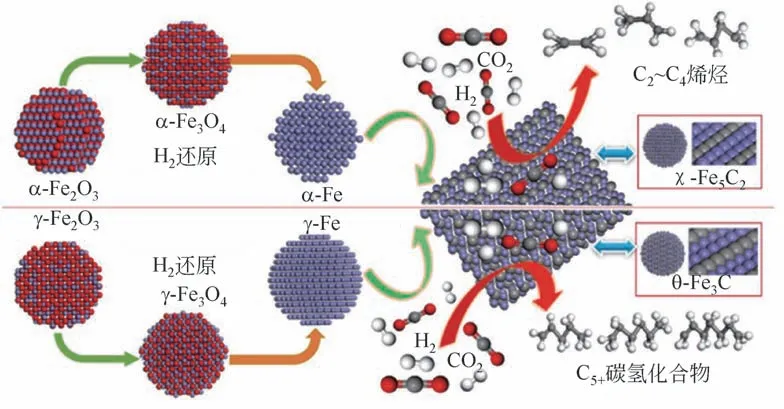
图20 α-Fe2O3和γ-Fe2O3在CO2加氢反应中的结构变化及构效关系[106]
图21 催化剂在反应器不同位置的失活原因[98]
张玉龙等[109]通过Operando Raman、in situ XRD 辅助其他离线表征技术(XPS、Mössbauer spectroscopy、TEM、FFT 等)研究了Fe 基催化剂全生命周期的构-效关系(活化-反应-失活-再生),发现活化后生成Fe5C2,在反应过程中会逐渐发生相转化,这是催化剂失活的主要原因(如图22),相转化的机理有两种(Fe5C2→Fe3O4和Fe5C2→Fe3C →Fe3O4)。 作 者 还 提 出 一 种CO2-CO 再生机理,可使活性及选择性均恢复90%以上。
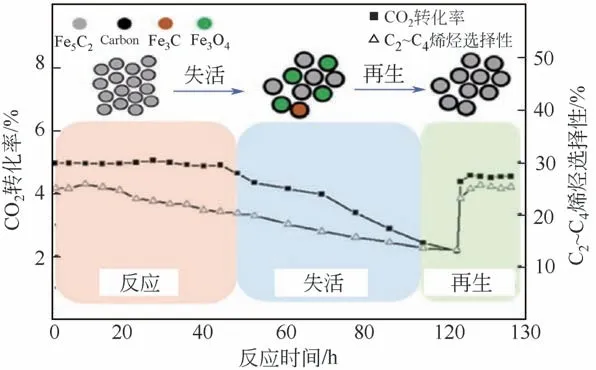
图22 CO2加氢全生命周期构效关系[109]
4 结语
CO2加氢制取烯烃可将CO2碳源转化为高附加值化学品,对于温室效应及减轻化石能源对外依存度具有重要战略意义。由于铁基催化剂对CO2加氢反应具有优异性能,其催化剂设计及改性、反应条件的筛选与优化、反应机理、构-效关系(活性相构成、分子模拟和动力学研究)等被广泛研究,但由于其活性相结构的复杂性及容易失活等难点,当前CO2加氢制取烯烃工艺尚未实现工业化,仍停留在基础研究的阶段,为尽快实现CO2加氢制取低碳烯烃的工业化目标,研发高性能催化剂是推动CO2加氢转化的关键。
在开发可应用于工业反应的高性能CO2加氢制取烯烃的Fe基催化剂的过程中,首先要在分子水平上准确认识催化剂的活性相组成、活性中心的形成过程以及可导致催化剂失活的诱因。通过研究催化剂在反应过程中的构-效关系,指导催化剂的设计和开发,不仅可以提高催化剂的研发效率,对其他工业催化剂开发也具有指导和借鉴意义。考虑到催化反应通常发生在材料的表界面,因而催化反应性能主要由参与反应的表界面结构性质决定。在工业反应过程中,尤其是类似于CO2加氢制取低碳烯烃这类高温、高压以及多种反应物共存的反应体系,催化剂结构往往会发生重构,甚至某些反应活性位只能在特定的反应环境下形成,随着反应时间或条件的变化可能还会导致催化剂失活。由此可见,在真实工业反应条件下,催化材料的表面结构具有显著的动态性。人们可以利用Operando技术对于反应的动态过程进行原位监测,特别是外界因素(如高温、高压等)所引起的催化材料表界面结构演化规律及作用机制,有望揭示其全生命周期内各种影响因素,为工业催化剂开发和设计提供方向。
猜你喜欢
炼油与化工(2022年4期)2022-10-10
科学家(2022年4期)2022-05-10
第一财经(2019年8期)2019-08-26
中南民族大学学报(自然科学版)(2019年1期)2019-04-04
科技创新与应用(2018年4期)2018-01-31
中学生数理化·高二版(2017年3期)2017-07-07
中学生数理化·高二版(2017年3期)2017-07-07
安徽医科大学学报(2015年9期)2015-12-16
汽车与新动力(2013年3期)2013-03-11
