
基于二代测序的RhD阳性个体RHD基因mRNA选择性剪接分析
2021-02-24 11:41罗亚林姬艳丽罗广平付涌水温机智黎诚耀
中山大学学报(医学科学版) 2021年1期
罗亚林,魏 玲,姬艳丽,罗广平,付涌水,,温机智,黎诚耀
(1.南方医科大学检验与生物技术学院输血医学系,广东广州 510515;2.广州血液中心临床输血研究所,广东广州 510095)
Rh是除ABO之外最重要的血型系统之一,其中D 抗原是Rh 血型系统中免疫原性最强的抗原,其相应同种抗体可引起严重溶血性输血反应和胎儿新生儿溶血病。编码D 抗原的RHD基因由10 个外显子组成,并与编码RhCE 抗原的RHCE基因具有高度同源性[1]。大部分人类基因在剪接过程中能够通过选择性剪接产生多种mRNA 转录本,使机体在不同组织和不同环境条件中能够利用一套固定的遗传物质表达出丰富的蛋白产物,以满足不同的生理需求[2]。前期研究显示,RHD基因也同样存在选择性剪接现象,这些选择性剪接形成的转录本能否表达出不同C-端的RhD 蛋白,且这些序列多样的RhD 蛋白是否有完整的D 抗原表位,迄今尚不清楚[3]。以往对RHD基因可变剪接的研究多采用逆转录聚合酶链反应(reverse transcription polymerase chain reaction,RT-PCR)扩增后分子克隆测序,或RT-PCR 后直接Sanger 测序的方法进行,对于低丰度的转录本难以检出,并且无法进行准确定量分析。本研究首先从RhD 阳性献血者的新鲜血液中分离出外周血单个核细胞(Peripheral blood mononuclear cell,PBMC),进行有核红细胞(Erythroblast)的体外扩增培养,提取红细胞特异性RNA,然后采用二代测序技术对RHD基因的cDNA 扩增产物进行测序分析,以期获得RHD基因选择性剪接转录本的定性和定量分析结果,并使用生物信息学方法对RHD基因选择性剪接转录本形成机制进行初步探讨。
1 材料与方法
1.1 研究对象
随机选取D 阳性献血者3 名,分别留取新鲜EDTA 抗凝外周静脉血10 mL。本研究经广州血液中心伦理委员会同意,并获得研究对象知情同意。
1.2 前体有核红细胞的分离与体外培养
参照我们已经建立的从外周血中分离、培养并扩增前体有核红细胞的方法[4]。采用Ficoll 密度梯度法分离PBMC,使用包含1 ng/mL 人白介素3(Interleukin-3,IL-3,Stem Cell 公司)、2 U/mL 促红细胞生成素(Erythropoietin,EPO,Stem Cell 公司)、1 μmol/L 地塞米松(Dexamethasone,Dex,Sigma 公司)、40 ng/mL 胰岛素样生长因子-1(Insulin-like growth factor-1,IGF-1,R&D 公司)、40 μg/mL cho⁃lesterol-rich lipids(Sigma 公司)和100 ng/mL 干细胞因子(Stem cell factor,SCF,Stem Cell 公司)的Stem⁃Span SFEM 培养基(Stem Cell 公司)进行筛选培养,调节细胞浓度至106/mL。在第5 天时使用Percoll分离液进行密度梯度离心,以纯化有核红细胞,并改用不含IL-3 的相同浓度细胞因子(即2 U/mL EPO、1 μmol/L Dex、40 ng/mLIGF-1、40 μg/mL cho⁃lesterol-richlipids 和100 ng/mL SCF)的StemSpan SFEM 培养基对有核红细胞进行培养,并将细胞浓度维持在1.5~2.0×106/mL[5]。
1.3 RNA的提取分离
采用QIA amp RNA Blood MiniKit 试剂盒(QIA⁃GEN GmbH,D-40724 Hilden,德国),按照试剂盒操作说明,提取有核红细胞总RNA,测定浓度分装后,置-80 ℃冰箱保存备用。
1.4 RT-PCR
依据逆转录试剂盒(PrimeScript,II 1st strand cDNA Synthesis kit,TaKaRa)操作说明,采用Oligo(dT)20 引物将RNA 反转录为cDNA。通过引物设计软件(Premer Premier 5.0)设计1 对特异性引物,扩增RHDmRNA的外显子6到3’非编码区,即正向引物位于外显子6(引物序列:5’-TGGCTGGGCT⁃GATCTCCG-3’),反向引物位于3’非编码区(引物序列:5’-TGCATAATAAATGGTGAGATTCTCCTC-3’)。随后,用GoTaq DNA 聚合酶(Promega 公司,USA)进行PCR 扩增。扩增条件为:94 ℃预变性5 min;然后94℃变性30 s,55℃退火30 s,72 ℃延伸1 min,共30个循环;最后72 ℃延伸10 min。PCR产物使用2%琼脂糖凝胶进行电泳分析。
1.5 PCR产物二代测序
将PCR 产物送至成都柯莱博生物科技有限公司,建库后使用Illumina Novaseq 测序仪进行二代测序。使用Hisat2-2.1.0 软件对测序结果和RHD基因组序列进行比对分析,测序结果采用StringTie v2.1.1 软件进行转录本拼接以及表达量计算,使用每千个碱基的转录和每百万映射读取的转录本(Transcripts Per Kilobase of exon model per Million mapped reads,TPM)对不同转录本相对表达量进行校正。
1.6 统计学方法
采 用MaxEntScan 软 件(http://hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html)对RHD基因各外显子的5’-剪接位点(5’-splice site,5’ss)和3’ss 保守区域序列进行生物信息学分析,计算反映5’ss和3’ss与剪接体结合力的指标,即最大熵值,以推测不同RHD转录本形成的原因。根据文献[6]报道,5’ss 区域包括外显子最后3 个碱基和内含子5’端的6个碱基,用该模型(Maximum En⁃tropy Model,MaxENT)计算的最理想保守序列最大熵值为11.81;3’ss 区域包括内含子3’端的6 个碱基和外显子的前3 个碱基,用该模型计算的最理想保守序列最大熵值为13.59。实际计算值越接近理想保守序列的最大熵值,表明在剪接时被剪接体识别且结合能力越强,更易被剪接。
2 结果
2.1 使用二代测序对RHD 基因mRNA 转录本进行分析
为了解RhD 阳性个体红细胞中RHD基因的各种转录本的表达情况,我们将RT-PCR 产物进行二代测序。测序结果表明:在RhD阳性标本中除了包含10 个外显子的RHD基因全长转录本外,还检测到了其它8 种不同的RHD基因mRNA 转录本(图1)。将结果进行校正后,每种转录本的相对表达量如图1所示。
2.2 生物信息学分析RHD 基因各外显子5’ss 与3’ss剪接区域剪接效能
对RhD 阳性个体各外显子5’ss 与3’ss 剪接区域的序列进行了生物信息学分析,计算了10 个外显子两端5’ss 的9 个碱基的保守序列(包括外显子最后3 个碱基和紧邻内含子的前六个碱基序列)和3’ss 的23 个碱基长度的保守序列(包括内含子的最后20 个碱基和紧邻外显子的前3 个碱基序列)的最大熵值(图2),计算结果显示RHD基因外显子7 的5’ss 保守区域最大熵值(2.93)和外显子2的3’ss保守区域最大熵值(2.83)明显低于最理想保守序列最大熵值,同时外显子7 和9 的3’ss 保守序列的最大熵值也相对较低(分别为6.98和6.15)。这些结果提示在RHD基因剪接过程中,剪接体与这几个外显子5’ss 剪接区域和3’ss 剪接区域的保守序列相互识别结合和结合能力下降,导致外显子2、7 和9 无法被正确识别,从而出现RHD基因被异常剪接形成相应外显子缺乏的转录本。

图1 三个RhD阳性个体中检出的不同RHD mRNA转录本及比例Fig.1 The expression frequencies of RHD mRNA transcripts in three RhD-positive individuals
此外,我们分析了位于IVS7+918_1087 区域(图2)的被异常插入RHD基因mRNA 转录本的170 bp 片段及其周围序列,发现其两端也存在GTAG保守序列,即其可能会被识别为一个“外显子”。生物信息学分析结果显示这170 bp 片段5’ss 序列的最大熵值为6.04,高于上游紧邻的外显子7 的5’ss 序列的最大熵值2.93(图2),所以在剪接过程中有可能被剪接体识别并作为一个外显子被剪接到成熟的mRNA转录本中。
3 讨论
本研究采用二代测序技术,对RhD阳性个体红细胞表达的RHD基因mRNA 转录本进行了分析,检测到了9种不同的RHD基因mRNA 转录本,其中包括3 种新的低丰度表达转录本(图1F-H)。自从Westhoff[7]等明确鉴定了第一个RHD基因转录本以来,多项研究在RhD 阳性个体红细胞中共发现了6 种不同的RHDmRNA 转录本[3],从脐血中提取的总RNA 中扩增检测到的RHDmRNA 转录本可高达13种,种类更加丰富[8]。以往研究在RhD阳性个体中报道过的转录本数目较少,这可能与这些研究采用一代Sanger 测序难以检出低丰度转录本有关。本研究首次采用二代测序技术对RHD基因RT-PCR 产物进行分析,一次能对几十万到几百万条DNA 分子进行序列测序,不但提高了检测的灵敏度,而且可以计算出每种转录本的相对表达量。虽然我们采用的测序方法较传统方法在检测水平上有了较大提高,但我们推测RhD 阳性个体可能仍然存在其它尚未发现的RHDmRNA转录本。
本研究仅扩增了RHD基因外显子6 到3’非编码区的序列(即RHD基因mRNA 后半部分)并进行了二代测序分析,而没有对RHD基因外显子1 至外显子10 进行全长扩增和二代测序分析。这是由于以往研究结果发现RHD基因的可变剪接主要集中在外显子7、外显子8 和外显子9[9]。此外,考虑到目前二代测序技术测序长度有限,仅扩增RHD基因mRNA 后半部分也可减少二代测序分析过程中序列拼接的次数,从而增加测序结果的可靠性。
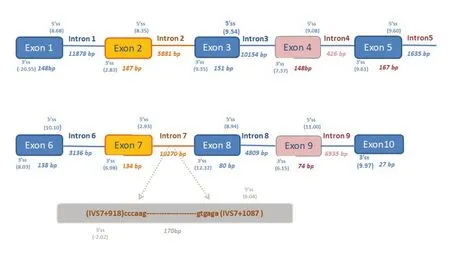
图2 RHD基因各外显子5’ss端和3’ss端最大熵值计算结果示意图Fig.2 The maximum entropy values of 5’and 3’splice sites of all exons of RHD gene
本研究中发现多种不同RHD基因mRNA 转录本,其变异形式存在外显子缺失以及内含子保留两种情况,具体表现为外显子7、外显子8和外显子9 存在不同组合的缺失,以及内含子7 部分序列(170 bp的片段)的保留,本研究发现了三种包含部分内含子7 序列(位于RHD基因IVS7+918_1087 区域)的RHD基因mRNA 转录本,这3 种转录本所占的比例均较低,其中3 种转录本(图1F、G 和H)发生移码突变,两种转录本提前出现终止密码子(图1G 和H)。此外,我们进一步对RHD基因这个内含子7 的170 bp 片段及其周围序列进行分析,发现这170 bp 的序列和既往报道的DEL 个体的异常插入序列一致[10]。另外,在长度为10 270 bp 的整个内含子7中,我们关注的经常被异常插入RHD基因mRNA 转录本的170 bp 片段的两端碱基序列符合剪接位点的GT-AG 原则。我们采用生物信息学方法模拟计算两端剪接序列的最大熵值,结果显示该170 bp 片段模拟的5’ss 剪接序列的最大熵值(6.04)明显大于RHD基因正常外显子7 的5’ss最大熵值(2.93)。因此我们推测在RHD基因剪接过程中,与5’ss 保守序列互补结合的U1 SNRNP(small nuclear ribonucleoproteins)剪接体,在识别外显子7 和内含子7 的170 bp 片段的5’ss 保守序列时,U1 SNRNP 剪接体与内含子7 的170 bp 片段的5’ss相似序列结合能力更强,可能会将这170 bp的内含子7 序列识别为一个“外显子”进行剪接,从而将这170 bp 的内含子7 序列保留在RHD基因mRNA中(图2)。
此外,以往不同研究得到的RHD基因mRNA转录本种类和数量上存在一定差异(图1),这可能是由于不同研究使用的红细胞种类不同。由于RhD 蛋白在红细胞中表达而不在白细胞中表达,但成熟红细胞已经脱核无法提取到RNA,故需要从尚未脱核的红细胞中提取RNA 进行RHD基因mRNA 转 录本 的分 析研 究。如Shao[11]等先 从新鲜全血中提取网织红细胞后再抽提RNA,然后再进行后续分析研究;许先国[8]等从脐带血中抽取总RNA 并进行后续分析研究。与以往研究不同,本研究中首次使用前期建立的有核红细胞的培养方法,从新鲜外周血分离、培养并扩增尚未脱核的有核红细胞,然后再提取RNA 并进行后续实验,检测到的常见转录本也与以往报道一致。与前期研究相比,本研究使用的方法能大量获得红细胞总RNA,提高了检测的灵敏度,并检测到3 种新的含量较少的RHD基因mRNA 转录本,该方法也可以应用于其他红细胞特异性表达基因的转录分析的研究中。
为分析外显子7和9容易发生剪接异常从而形成外显子7 和9 缺失转录本的原因,我们对RHD基因所有外显子的5’ss 和3’ss 保守区域序列进行了生物信息学分析。5’ss 和3’ss 剪接保守序列位于内含子和外显子的交界处,是在进化过程中形成的相对保守区域,至少包括5’ss 的9 个碱基及3’ss 的23 个碱基序列,该区域中最保守的是内含子的5’GT 和3’AG 位点,但也存在一定的变异。MaxENT模型可对该保守区域进行分析,5’ss和3’ss区域最理想的保守序列的最大熵值分别为11.81 和13.59。如果一个基因某个外显子剪接序列的数值越接近最大值,即表明其序列为一个真正的剪接位点几率越大,更易被剪接体识别而发生剪接。分析结果显示外显子7 的5’ss 序列最大熵值相对于其他外显子较低,即外显子7 的5’ss 序列在剪接起始阶段与U1 snRNP 剪接体的相互结合能力可能出现显著下降,导致其不易被U1 snRNP 剪接体识别并结合,从而发生异常剪接。此外,外显子9 的5’ss 序列最大熵值正常,仅存在3’ss 序列最大熵值中度下降的情况,外显子9 异常剪接是否由3’ss 与其互补结合的U2 snRNP 剪接体结合减弱相关,还需要进一步的研究。
总之,RHD基因存在十分复杂的选择性剪接模式,使RhD 阳性个体的RHD基因在mRNA 水平上存在多种不同的剪接转录本。本研究首次采用二代测序技术,检测到RhD 阳性个体红细胞9 种不同的RHD基因mRNA 转录本,其中包括三种新的低丰度转录本,从而为进一步了解RhD 蛋白的抗原表位及其在临床同种免疫中的意义奠定了基础,亦将为探讨剪接异常所致RhD 变异型的分子机制研究提供帮助。这些不同的RHD基因转录本的形成机制以及其能否翻译出含正常D 抗原表位的RhD 蛋白,目前尚不清楚,仍需进一步研究。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
南京医科大学学报(自然科学版)(2021年8期)2021-10-19
科学之谜(2021年2期)2021-04-25
中国生殖健康(2020年4期)2021-01-18
教学考试(高考生物)(2020年6期)2020-11-23
山地农业生物学报(2020年2期)2020-11-09
现代农业科技(2020年15期)2020-08-16
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
湖北农业科学(2014年11期)2014-09-10
