
血管紧张素-Ⅱ与急性胰腺炎
2021-02-07 01:50黄子俊吕永才雷静静
世界华人消化杂志 2021年1期
黄子俊,吕永才,雷静静,刘 琦
黄子俊, 贵州医科大学 贵州省贵阳市 550004
吕永才,镇宁布依族苗族自治县人民医院消化内科 贵州省镇宁县561200
雷静静, 贵州医科大学附属白云医院消化内科 贵州省贵阳市 550014
刘琦,贵州医科大学附属医院消化内科 贵州省贵阳市 550004
0 引言
急性胰腺炎(acute pancreatitis,AP)是多种病因导致胰酶在胰腺内被激活后引起胰腺组织的自身消化、水肿、出血甚至坏死,伴或不伴有其他器官功能改变的炎症性疾病,临床上大多数患者的病程可呈自限性,AP的总死亡率约5%-10%[1-5],其中SAP的死亡率约30%-40%[6,7].根据2012年亚特兰大分类标准修订版本[8],并根据AP的严重程度将其分为轻症急性胰腺炎(mild acute pancreatitis,MAP)、中重症急性胰腺炎(moderately severe acute pancreatitis,MSAP)及重症急性胰腺炎(severe acute pancreatitis,SAP),其中约60%病例是MAP,约30%的患者会发生MSAP,约10%会发生SAP,持续器官衰竭(persistent organ failure,POF)是AP患者早期死亡的主要原因,胰腺坏死继发感染是后期AP患者的主要死因,死亡率极高[9-11].AP病情进展及预后与疾病的早期治疗及干预息息相关,而AP的发病机制的复杂性又影响着疾病的早期治疗的疗效,但目前AP的发病机制尚未完全阐明,故缺乏特异性治疗,现今公认发病机制有“白细胞过度激活—炎性因子级联瀑布学说”[12]、“肠道细菌移位与二次打击学说”[13]、“细胞凋亡学说”[12]、“胰腺微循环障碍学说”[14]等,这些理论的提出表明AP的发病机制是复杂、多因素参与的病理生理过程.近年来的研究表明,肾素-血管紧张素系统(reninangiotensin system,RAS)中的活性物质-血管紧张素Ⅱ(angiotensin-Ⅱ,Ang-Ⅱ)在AP的发生发展中起着至关重要的作用(图1)[15-17].
1 Ang-Ⅱ概述
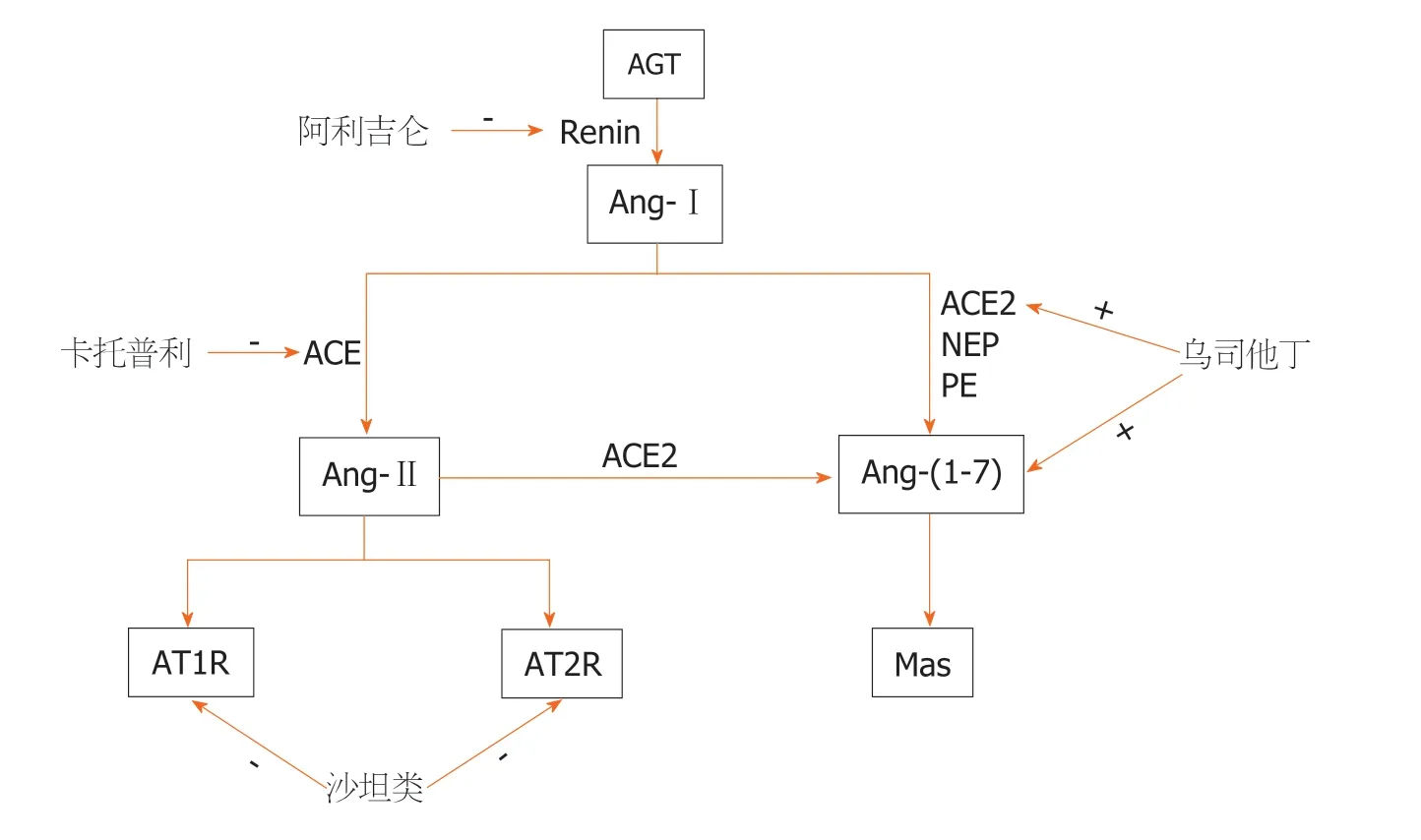
图1 针对血管紧张素Ⅱ活性各位点的急性胰腺炎治疗方法示意图. Renin:肾素; AGT:肾素血管紧张素原; Ang-Ⅰ:血管紧张素Ⅰ; Ang-Ⅱ:血管紧张素Ⅱ; ACE:血管紧张素转换酶; ACE:血管紧张素转换酶2; AT1R:血管紧张素Ⅱ受体1; AT2R:血管紧张素Ⅱ受体2; Ang(1-7):血管紧张素(1-7).
Ang-Ⅱ是由8个氨基酸组成的线状小肽,它是RAS中最主要的生物学活性物质之一,主要由血管紧张素Ⅰ(angiotensin-Ⅰ,Ang-Ⅰ)在血管紧张素转换酶(angiotensin converting enzyme,ACE)的作用下生成,并通过与血管紧张素Ⅱ受体1(Ang-Ⅱ type1 receptor,AT1R)、血管紧张素Ⅱ受体2(Ang-Ⅱ type2 receptor,AT2R)、血管紧张素Ⅱ受体3(Ang-Ⅱ type3 receptor,AT3R)和血管紧张素Ⅱ受体4(Ang-Ⅱ type4 receptor,AT4R)结合发挥作用[17,18].一般认为Ang-Ⅱ与AT1R结合会导致血管收缩、氧化应激和炎症介质释放等,而刺激AT2R时,其效应与AT1R相对,与AT3R和AT4R结合的功能尚不清楚.目前有学者认为当激动AT3R时,可能与血管收缩有一定联系,而激动AT4R可能与细胞记忆、回忆有关,同时也可能影响血流动力学和钠潴留,但具体机制尚不清楚[15].Ang-Ⅱ还可在氨基肽酶A的作用下通过其N末端切割形成七肽的血管紧张素-Ⅲ(angiotensin-Ⅲ,Ang-Ⅲ),Ang-Ⅲ与多种血管紧张素受体(包括AT1、AT2和AT3受体)相互作用而与Ang-Ⅱ相似,但作用机制较Ang-II弱[18,19]; 且Ang-Ⅱ在ACE2的作用下形成血管紧张素(1-7)[angiotensin(1-7),Ang(1-7)],但Ang(1-7)主要由Ang-Ⅰ在ACE2的作用形成,即组成新轴ACE2-Ang(1-7)-Mas,并拮抗经典轴ACE-Ang-Ⅱ-AT1,认为新轴对AP具有潜在的保护作用[20,21].
2 Ang-Ⅱ在循环RAS中的生成
AP是一种炎症反应疾病,会使机体持续处于应激状态,RAS主要由炎症级联反应激活[16,22].循环RAS激活后,肾小球系膜细胞分泌肾素,将肝脏合成的血管紧张素原转化为Ang-Ⅰ,随后ACE在肺循环及其他血管内皮细胞中产生,将Ang-Ⅰ转化为Ang-Ⅱ,同时在其他一些酶的作用下Ang-Ⅱ还可以产生其他一些活性物质,如Ang-Ⅲ,Ang(1-7)等,这是经典ACE介导的Ang-Ⅱ生成途径.除上述经典途径外,研究表明循环RAS中可能至少还有两条Ang-Ⅱ产生的途径[12]:(1)直接由激肽释放酶,组织蛋白酶G等催化Ang-Ⅰ转换为Ang-Ⅱ; (2)通过组织纤溶酶原激活物、组织蛋白酶G等酶作用下将血管紧张素原催化形成Ang-Ⅱ[23,24].因此,当循环RAS被激活后,至少将沿着上述三个路径生成许多Ang-Ⅱ.
3 Ang-Ⅱ在胰腺局部的表达
在1991年Chappell等[25]首次在犬的胰腺组织中检测到Ang-Ⅱ、血管紧张素原,且发现胰腺局部Ang-Ⅱ的浓度高于外周血Ang-Ⅱ的浓度,表明犬的胰腺组织中存在局部Ang-Ⅱ的生成,随后研究中再次首次报道大鼠胰腺腺泡AR42J细胞系表达ACE-Ang-Ⅱ-AT1R轴的全部组分,包括:肾素、血管紧张素原、ACE和AT1aR、AT1bR、AT2R; 在1997年Leung等[26]报告了大鼠的胰腺中存在局部RAS,并且发现AT1和AT2受体的表达主要在血管内皮和胰腺导管系统的上皮中,而在腺泡中浓度较低,后来该小组应用蛋白质印记法、半定量反转录-PCR以及免疫组化等实验方法证实在大鼠实验性AP中胰腺局部RAS相关基因及合成蛋白表达增加; 随后Tahmasebi等[27]在人的胰腺组织局部也发现RAS存在,并发现RAS通过旁分泌产生的Ang-Ⅱ可能直接影响胰腺局部血流和胰岛B细胞功能,故认为Ang-Ⅱ在调节胰腺内分泌、外分泌功能方面可能有重要作用.综上这些发现支持胰腺存在完整RAS,其作用主要由Ang-Ⅱ介导.
4 Ang-Ⅱ在AP中的作用机制
4.1 Ang-Ⅱ与胰腺微循环障碍 AP为炎症性疾病,炎症反应激活RAS,进一步产生其重要的活性产物Ang-Ⅱ,且Ang-Ⅱ、AT1R的表达主要在血管内皮和胰腺导管上皮细胞中,Ang-Ⅱ与AT1R结合,将引起胰腺的微动脉收缩,使胰腺发生缺血、缺氧,最终导致胰腺发生血管收缩、毛细血管淤血和局部贫血,反过来Ang-Ⅱ与AT1R结合后引起炎症因子如白介素-6、肿瘤坏死因子-α、白介素-10等的释放,导致该过程恶性循环[28-31].Pan等[31]研究表明大鼠模型的AP中发现缬沙坦通过阻断Ang-Ⅱ与AT1R结合抑制胰腺微循环障碍和炎症反应,使胰蛋白酶活性降低,从而使AP的病情得到改善.陈强等[32]使用高渗盐水也证实通过抑制醛固酮、Ang-Ⅱ的分泌,对AP引起的微循环障碍有改善作用.
4.2 Ang-Ⅱ与促炎症介质 在AP的炎症应答过程中,致炎因子和氧化应激触发共同的信号传导通路,主要通过丝裂原活化蛋白激酶/核因子激活,导致炎症的级联扩增,进而加重AP的进展[15,33,34].而致炎因子和氧化应激触发在RAS激活途径中主要由Ang-Ⅱ与AT1R结合介导.汤伟胜等[35]与任勇等[36]最近研究表明AP患者血清中白介素-6、白介素-8、白介素-10、CRP、肿瘤坏死因子-α等炎症介质含量较正常人高,且AP患者血清Ang-Ⅱ的浓度与血清白介素-6、白介素-8、白介素-10、C-反应蛋白(C-reactive protein,CRP)、肿瘤坏死因子-α浓度呈正相关,证实了RAS激活是导致全身炎症反应持续进展的重要因素.
4.3 Ang-Ⅱ与氧自由基 AT1受体是一种G蛋白偶联受体,通过激活磷脂酶A、磷脂酶C、磷脂酶D或蛋白激酶C而引起作用,在胰腺组织中广泛存在,当Ang-Ⅱ与AT1受体结合时,通过激活烟酰胺腺嘌呤二核苷酸磷酸氧化酶(reduced form of nicotinamide-adenine dinucleotide phosphate,NADH/NADPH)产生活性氧(reactive oxygen species,ROS),ROS诱导的氧自由基释放最近在某些病理条件下已被证实,包括AP,氧自由基会对胰腺组织进行攻击,导致胰腺坏死[15,17].一项研究使用夹竹桃麻素(一种强效的NADPH抑制剂)的研究发现ROS蛋白生成明显受到抑制,说明Ang-Ⅱ诱导的氧自由基相关疾病如AP由NADPH氧化酶介导,同时促进细胞因子的活化并产生大量氧自由基,引起细胞坏死或凋亡,造成胰腺及胰腺外脏器的功能障碍甚至衰竭[37].值得注意的是,应用血管紧张素受体拮抗剂(angiotensin receptor blocker,ARB)可以减轻ROS的产生和随后的氧化应激,已经成为在多种疾病中进行氧化应激管理的良好候选药,但在AP临床应用中暂未得到推广.
研究者们还发现黄嘌呤氧化酶(xanthine oxidase,XOD)和线粒体中ROS,也能致使Ang-Ⅱ诱导NADPH氧化酶激活,产生大量的氧自由基[15].在一项高血压合并痛风(痛风患者服用黄嘌呤氧化酶抑制剂)研究中发现与单纯高血压患者相比,学者们发现前者血管内皮活性减低,如血管内皮生长因子浓度减少[37].线粒体已经被证明为真核细胞中ROS的来源,线粒体中ROS产生由线粒体ATP敏感钾通道介导,研究者们使用鱼藤酮(ATP敏感钾通道抑制剂)抑制线粒体膜去极化时,在牛的主动脉内皮细胞中,也观察到了P物质、血管内皮生长因子合成减少[38].综上分析认为Ang-Ⅱ致使的NADPH氧化酶激活所致氧自由基的产生也由XOD和线粒体中ROS来激活,虽未在AP的研究中进行相关报道研究,但也许为未来AP治疗提供新的理论依据.
4.4 Ang-Ⅱ与胃肠激素 研究认为胰腺局部RAS激活与其他胃肠激素分泌存在负反馈调节的机制,故胰腺局部RAS激活使胰液分泌减少,进而反馈引起胃肠激素大量分泌,如缩胆囊素或其他胃肠激素,刺激已损伤的胰腺继续分泌胰酶,加重AP病情[39].
5 Ang-Ⅱ相关拮抗剂在AP中的运用现状
5.1 肾素抑制剂 肾素作为RAS级联反应首环节限速酶,对Ang-Ⅱ的生成起重要调控作用.阿利吉仑是直接肾素抑制剂,对肾素具有很高的亲和力,能与肾素活性位点结合阻断其催化活性.从RAS的源头阻断Ang-Ⅰ、Ang-Ⅱ的产生,目前阿利吉仑被批准用于治疗高血压,其对AP的炎症病变及损伤也具有一定保护作用.黄元龙等[40]在大鼠AP模型中通过阿利吉伦(肾素抑制剂)抑制Ang-Ⅱ的生成,抑制了核因子-κB、信号转导通路,从而抑制肿瘤坏死因子-α生成,使得胰腺水肿、出血、坏死得到改善,故认为阿利吉伦通过抗炎作用AP进展.
5.2 ACE抑制剂 ACE抑制剂能抑制Ang-Ⅰ向Ang-Ⅱ转化,对AP具有潜在的治疗作用,ACE抑制剂卡托普利预处理,可降低Ang-Ⅱ生成和AT1R表达,抑制Rho相关蛋白激(Rho-associated protein kinase,Rho/ROCK)通路,降低胰和肺组织病学评分,保护AP及相关的肺损伤,但卡托普利预处理后,只有ROCK2的表达急剧下降,而ROCK1的表达略有下降[41].El-Ashmawy等[28]再次证明,用卡托普利(Captopril,CAP)和甲基强的松龙(Methylprednisolone,MP)预处理均显示胰腺细胞组织病理学改变的改善,如胰腺水肿减轻,腺泡细胞变性改善,炎性细胞渗透减少,这些结果与以往的研究一致.目前血管水肿是这类药物公认的不良反应,据报道ACEI引起的血管水肿的发生率从0.1%到1.0%不等.ACEI血管水肿是一类效应,并不是剂量依赖性的,因此,症状可在最初剂量后几小时至10年内随时发生.Gorsane等[42]在一个临床案例中也发现这一点,他们在停用卡托普利后,血管性水肿减轻.ACEI血管水肿的病理生理学仍有争议,钙激活钾通道亚基α-1的共同变异被认为与ACEI或ARB治疗引起的血管水肿的风险有关[43],故ACE抑制剂的使用可能诱发AP.
5.3 AT1受体拮抗剂 ARB包括AT1R、AT2R拮抗剂(沙坦类)等.ARB拮抗AT1R在RAS末端的受体水平抑制,由AT1R介导的细胞内NADH/NADPH氧化酶的活性,从而减少ROS的产生.AT1受体阻制剂缬沙坦是一种非肽竞争性拮抗剂,能高度选择性阻断AT1受体,从而导致Ang-Ⅱ的不利影响.缬沙坦因其安全性高、疗效高、耐受性高、作用时间长、副作用小等优点,广泛应用于治疗和预防高血压、冠心病等心血管疾病.Pan等[31]在大鼠模型的AP中发现缬沙坦通过阻断Ang-Ⅱ与AT1R结合抑制胰腺微循环障碍和炎症反应,使胰蛋白酶活性降低,从而使AP的病情得到改善.一项基于瑞典人群的病例对照研究表明,血管紧张素-Ⅱ受体阻制剂对AP的风险有保护作用[30].Bostanci等[5]的实验表明,坎地沙坦可通过改善AP中的胰腺微循环障碍来减轻胰腺组织水肿、炎症和腺泡细胞坏死,还证明坎地沙坦可降低胰腺组织明胶酶B的表达,抑制明胶酶B的表达可能导致AP中粒细胞和炎症反应的减少.
5.4 其他 Yang等[45]研究证明Ang-Ⅱ预处理的人类脐带间充质干细胞通过抑制炎症、减少胰腺损伤和促进SAP中的胰腺血管生成,这种新的靶向治疗可能有利于SAP的治疗.乌司他丁为一种蛋白酶抑制剂,是临床使用最早的抗自由基药物之一,可稳定溶酶体膜,抑制溶酶体酶的释放,在治疗AP过程中有举足轻重的作用,产生作用主要是通过上调Ang-(1-7)和ACE2的表达来重建RAS的平衡,从而改善细胞凋亡,显著改善胰腺病理,可能也具有AP临床治疗潜力[46].血液净化可有效清除血液中的细胞因子、炎症介质、内毒素等中小分子毒性物质和代谢产物,调节水、电解质和酸碱平衡,有助于改善机体各重要器官功能,避免多器官功能障碍的发生,对SAP患者进行连续性血液净化治疗,其炎性指标及 RAS指标明显下降,其在改善RAS和炎症因子激活以及疗效方面均优于基础治疗[47-49].研究表明在AP发病72 h内行连续性血液净化可以尽早干预并控制炎症的级联放大反应,乌司他丁联合早期连续静脉-静脉血液滤过模式序贯治疗SAP可发挥协同治疗作用,降低由此造成的各脏器损害,有效抑制酶促反应和全身炎性级联反应,调整炎症反应平衡,改善血清炎性细胞因子水平,促进患者症状及体征缓解,预防局部并发症,缩短病程,改善预后[50].
6 Ang-Ⅱ与遗传性AP的关系
遗传性胰腺炎是指发生在20岁以下并至少有两位近亲发生胰腺炎的病人[51].近几十年来,将遗传多态性与炎症性疾病相关的研究引起了广泛关注,当然包括AP.该领域研究结果表明应该对易患胰腺炎(包括AP、慢性胰腺炎)的个体进行基因改变筛查,以便于分娩时进行预防措施.随着miRs(一种小型的非编码RNA)和其他表观遗传控制机制的发现,影响miR结合的RAS基因多态性(或转录靶向RAS基因的miR基因位点的遗传变异)可能是决定个体患者胰腺炎易感性的关键因素,而在其中有学者提出RAS中活性产物升高可能起着重要诊断临床意义,其中包括活性产物Ang-Ⅰ、Ang-Ⅱ及相关转换酶等[15],但由于该方面研究费用昂贵导致该领域的报道少,目前临床研究与基础研究结果存在争议,且机制尚不清楚,目前在临床上仍难以推行.故以后应加大该领域研究,可能为临床罕见病因或是特发性AP提供治疗依据.
7 Ang-Ⅱ与AP的严重程度
Ang-Ⅱ为RAS系统最重要的活性产物,而RAS是一个调节机体血压及水电解质平衡的重要系统,其主要激活途径是依赖于多酶促级联反应.其中大量基础文献证实其在AP的病理生理作用,但针对RAS在AP患者中,是否一定浓度Ang-Ⅱ能预测SAP、多器官功能衰竭、感染性并发症、肠缺血以及死亡率仍是缺乏的,未来仍需大量研究进行证实.
8 问题与展望
AP中Ang-Ⅱ生成及它在AP病情演变中的作用机制目前还存在许多问题待阐明.首先,循环中Ang-Ⅱ和胰腺局部组织Ang-Ⅱ的生成之间是否存在一定关系及其具体有何联系仍未阐明.其次,通过何种途径阻断AP中Ang-Ⅱ活性对于AP的实验及临床治疗效果,同时其与早期AP严重程度相关,仍需大量的研究来证实.再者,Ang-Ⅱ在AP中作为一个炎症反应前的因子具体通过何种途径调控其下游炎性因子的表达仍需深入探索,且AP患者中Ang-Ⅱ与AT1R结合后由NADPH引起细胞信号传导所致氧自由基释放与线粒体氧化物和黄嘌呤氧化物介导氧自由基之间是如何调节.最后,遗传性AP的研究在基因水平上是否可通过基因的表观遗传修饰得以控制可能成为未来研究热点.综上所述,对于AP中RAS激活及其最重要的活性产物Ang-Ⅱ水平异常升高的进一步研究将有助于加深对AP发病机制的认识,并有助于从新的角度解释AP的发病机制奠定理论基础.而以Ang-Ⅱ介导NADPH氧化酶起细胞信号传导所致氧自由基为靶点的各种药物及基因修饰未来可能成为深入研究AP的发病机制提供新的工具,还可能对实验和临床治疗AP有重要的指导.
猜你喜欢
中国典型病例大全(2022年7期)2022-04-22
现代仪器与医疗(2021年6期)2022-01-18
肝胆胰外科杂志(2021年10期)2021-11-18
健康之家(2021年6期)2021-09-08
世界睡眠医学杂志(2021年4期)2021-07-03
智慧健康(2021年33期)2021-03-16
人人健康(2020年4期)2020-05-25
Food and Health(2019年2期)2019-05-17
中国医药科学(2016年8期)2016-10-09
中国实用医药(2016年9期)2016-05-17
