
Rab蛋白家族在神经类疾病中的作用
2021-02-02 08:28吴安平庆宏全贞贞
遗传 2021年1期
吴安平,庆宏,全贞贞
Rab蛋白家族在神经类疾病中的作用
吴安平,庆宏,全贞贞
北京理工大学生命学院分子医学和生物诊疗工信部重点实验室,北京 100081
细胞内膜囊泡运输是一个复杂的通路网络,Rab GTPases是膜囊泡运输的主要调节剂,通常被认为是细胞内吞和分泌系统中各种细胞器和囊泡的特异性标记和识别物。与Rab蛋白相关的轴突运输、内体运输发生障碍是造成神经退行性疾病的重要原因之一。本文主要介绍了Rab蛋白在多种神经退行性疾病病理机制中的作用机理与调控机制,同时讨论了线粒体和胶质细胞功能异常与Rab蛋白之间的关联。深入探究Rab蛋白的作用机制对人类神经性疾病的早期诊断和治疗具有潜在的指导意义。
Rab蛋白;神经退行性疾病;膜囊泡运输;线粒体;星型胶质细胞
中国是世界上痴呆症患者最多的国家,给公共和卫生保健系统带来了沉重负担。研究表明,2016年我国老年人口患痴呆症的概率是5.6%[1],目前我国的痴呆症患者大约有3100万,给患者及其家人带来沉重负担,将会引发越来越严重的公共卫生问题和社会问题[2]。
痴呆等认知障碍的形成是由于神经元结构和功能逐渐丧失,以及神经元死亡和胶质细胞失衡所导致的,主要包括阿尔兹海默病(Alzheimer’s disease, AD)、帕金森病(Parkinson’s disease, PD)、亨廷顿病(Huntington’s disease, HD)、肌萎缩性侧索硬化症(amyotrophic lateral sclerosis, ALS)、不同类型脊髓小脑共济失调病等神经类疾病。研究表明Rab蛋白能够调节神经元细胞的内吞和轴突运输,参与多种神经类疾病的病理进程。本文就Rab蛋白如何调控各类神经类疾病进行了归纳和分析,进一步阐述了Rab蛋白在的作用机制,为今后探究Rab蛋白在神经类疾病中的作用提供了思路和参考。
1 Rab蛋白结构
Rab蛋白是存在于细胞质膜和细胞器膜中的一类调节型小分子GTP结合蛋白,是Ras超家族中最大的亚家族。Rab蛋白在进化上高度保守,几乎存在于所有的真核生物中。Rab蛋白平均分子量大小为25 kDa,约由200个氨基酸组成。Rab蛋白含G结构域、N端与C端,其中,G结构域高度保守,N端与C端高度可变[3]。G结构域包含6个β折叠、5个α螺旋及5个多肽环,多肽环的氨基酸序列高度保守,是Mg2+和鸟嘌呤的结合位点,同时催化GTP水解。Rab蛋白的羧基末端包含-XXXCC-、-XXCCX-、-XCCXX-、-CCXXX-或-XXCXC-基本序列,其中两个半胱氨酸是异戊二烯化的底物。这种修饰对于膜结合至关重要。羧基末端高变区是必需的,但不足以将Rab蛋白正确靶向到细胞中的特定位置[3]。大多数Rab蛋白通过翻译后在羧基端附近的两个半胱氨酸上将两个香叶基(20碳聚异戊二烯基)基团与膜或膜蛋白复合物紧密结合,少数Rab蛋白只有一个香叶基[4]。N端的作用可能是参与指导C端半胱氨酸进行异戊二烯化修饰。G结构域的“开关”区域(称为开关1和2)通过与GTP结合产生稳定的构象,是Rab蛋白特异性识别其细胞效应子关键区域。不同的Rab蛋白在进行开“ON”和关“OFF”状态切换时,可能会发生比较灵活的构象变化[5]。对Rab蛋白突变体及嵌合体的研究表明,分子开关I和分子开关II、高度可变的N端和C端是Rab蛋白重要功能的决定因素。
Rab蛋白的主要功能是调节细胞信号的传导、细胞的生长和分化[6]。Rab GTPases作为分子开关,在GTP结合的活性形式和GDP结合的非活性形式之间转换,控制细胞内囊泡运输的各个方面。Rab-GTP结合型和Rab-GDP结合型之间的循环受蛋白质调节剂严格控制:GDP解离抑制剂(GDP dissociation inhibitors, GDI)调控Rab蛋白与膜的连接及Rab蛋白从膜上解离,GDI作为循环因子起作用,只与异戊二烯化修饰的无活性的Rab蛋白结合,将Rab-GDP保持在胞液中。伴随着Rab蛋白与供体囊泡的膜或膜蛋白复合物结合,鸟嘌呤交换因子(guanine nucleotide exchange factor, GEF)催化Rab- GDP形式转化成Rab-GTP形式,Rab蛋白被激活,Rab-GTP与下游效应子相互作用,使效应子功能发生变化;GTPase活化蛋白(GTPase accelerating protein, GAP)催化GTP水解,Rab蛋白失活,不再与效应子相互作用;GDI从膜上抽出Rab-GDP,与其形成复合物,又循环到胞液中。通过激活/失活循环,Rab蛋白作为分子开关将上游信号传递给下游效应子(图1)。Rab蛋白在其活性形式时与许多不同的蛋白质相互作用,这些蛋白质被称为Rab蛋白的效应子。Rab蛋白的效应子在囊泡的形成、囊泡沿细胞骨架的转运及囊泡与靶膜的拴系锚定等阶段中发挥作用[7]。
2 Rab蛋白在细胞中的调控途径
Rab蛋白最主要的功能是调控细胞内吞,内吞运输系统主要由内吞、循环、降解等子系统组成。其中,内吞循环运输负责将膜上大分子送回质膜回收再利用,在细胞极性形成和维持、细胞分裂及迁移、神经突触可塑性、免疫应答、生长因子受体调控等过程中不可或缺[8]。降解途径导向晚期内含体和溶酶体,货物在其中发生降解[9]。沿内吞途径的每个运输步骤均由不同的Rab蛋白介导:Rab5介导货物从质膜到早期内含体的运输并充当早期内含体的标记物[10];而Rab7则调节货物从早期到晚期内含体的运输并充当晚期内含体的标记物[11];Rab4,Rab11和Rab35介导回收途径;Rab4和Rab35调控货物从早期内含体、循环内含体直接返回质膜的快速回收[12,13];Rab11控制循环内含体的回收[14](图2)。研究表明,晚期内含体和溶酶体功能异常,在细胞衰老和神经退行性疾病(如帕金森病和阿尔茨海默病)中起着关键作用。
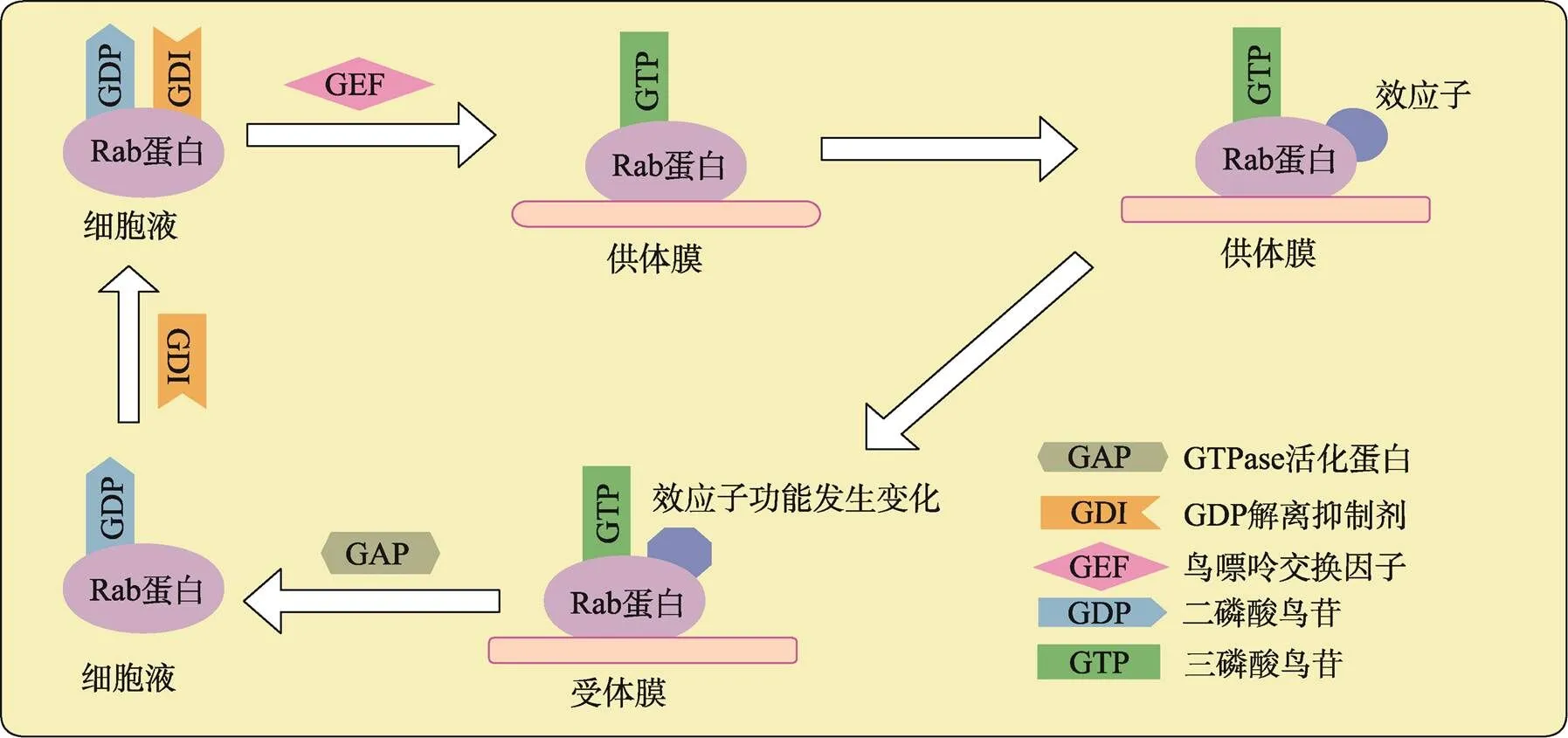
图1 Rab蛋白的功能循环示意图
Rab蛋白与GDI、GDP结合保存在胞液中,由GEF激活转移到供体膜上转化成Rab-GTP形式,使下游效应子发挥作用,激活态的Rab-GTP蛋白被GAP催化水解,随后GDI与Rab-GDP又形成复合物,循环到胞液中,形成循环。

图2 Rab蛋白在细胞中的分布
ER:内质网;TGN: 反高尔基网络;EE:早期内含体;LE:晚期内含体;RE:循环内含体;AP:自噬体;ERGIC:内质网一高尔基体中介组分;L/V:溶酶体/液泡;PAS:前自噬体;SV:分泌囊泡/颗粒。
3 Rab蛋白在神经元中的相关功能
神经元的特殊形态和功能高度依赖于膜运输的严格调节。内吞功能障碍破坏了双向轴突运输和突触小泡与靶膜的对接,从而导致神经运输和神经营养信号发生异常。轴突运输的改变,特别是神经营养蛋白受体在轴突运输的改变与几种人类神经退行性疾病都有关联[15,16]。Rab3A是突触活性区的结构成分,与其他核心活性区蛋白RIM1和MUNC13一起形成三元复合物,调节突触小泡与膜的结合过程,以释放神经递质[17]。Rab3蛋白还参与淀粉样前体蛋白(amyloid protein precursor, APP)的快速轴突运输[18]。Rab7控制神经营养蛋白受体的逆行轴突运输[19],Rab7损伤可诱导神经营养因子TrkA在内含体中的积累,进而阻滞神经营养蛋白逆行转运[20]。Rab26位于突触小泡表面,自噬蛋白Atg16L1、LC3和Rab33B被募集到这些突触小泡中,从而连接了突触小泡与自噬机制[21]。与其他细胞类型相比,神经元可能对蛋白质水平的轻微升高或降低更敏感,特定蛋白质水平的微小变化也可能导致神经元功能障碍和死亡。
4 Rab蛋白在神经退行性疾病中的作用
4.1 阿尔茨海默病
阿尔茨海默病是神经变性和痴呆症的最常见形式。超过90%~95%的AD病例是散发性(sporadic AD,SAD)且与已知的疾病突变无关。大约5%~10%的病例被归类为家族性AD (familial AD, FAD),与早老素1、早老素2和APP的突变有关。这些突变导致β-淀粉样蛋白(β-amyloid, Aβ)的产生发生异常。在Aβ形成途径中,APP首先被β-分泌酶切割,其中主要发挥作用的是β-分泌酶1 (β-site APP cleaving enzyme 1, BACE1),使APP 596~597位氨基酸之间发生裂解,释放一个可溶性的约100 kDa的N端片段sAPPβ,在膜上留下一个12 kDa的C端片段C99,C99片段被γ-分泌酶切割,从而产生Aβ。AD患者中Aβ产生过量,作为细胞外沉积物积聚在脑中,是形成老年斑的主要成分,其与由tau蛋白异常磷酸化导致的神经原纤维缠结是AD最主要的两大病理特征[22]。
Rab11b位于早期和晚期内体,高尔基复合体和内质网中。通常与再循环内含体有关,调控着反面高尔基体管网状结构与内吞、在循环内含体或质膜之间的物质运输。通过共聚焦分析和免疫荧光染色显示,在Rab11B阳性循环内含体中观察到BACE1沿树突和轴突定位并运输。沉默导致内在的BACE1在体细胞中积累,伴随着轴突中BACE1的水平降低[23]。这些结果表明,Rab11是神经元中BACE1的分选调节剂,对于BACE1的轴突分选至关重要。另外,siRNA敲低的表达显著降低了sAPPβ和Aβ的水平,这提示Rab11功能障碍可能是部分散发性阿尔茨海默病病例的发病机制之一[24]。Rab21在蛋白从早期内含体向晚期内含体转运过程中发挥作用,研究发现过表达可以通过促进γ-分泌酶的内吞及向晚期内含体的迁移来影响其活性,进而促进Aβ的产生[25]。沉默会降低Aβ水平,而不影响sAPPβ。这意味着这些Rab GTPases会影响γ-分泌酶对APP的水解或Aβ的运输[25,26]。同济大学生命科学与技术学院的裴刚院士课题组通过采用双分子荧光互补技术结合荧光共振能量转移技术发现ADAM10/BACE1二元复合物主要位于早期内含体,进一步观察到γ-分泌酶与α-/β-分泌酶二元复合物相互作用,表明α-、β-和γ-分泌酶可能形成三元复合物[27],这提示影响γ-分泌酶的Rab21蛋白可能同时调控α-分泌酶和β-分泌酶。Rab8A/Rab8是极化运输的重要调节剂,并参与反高尔基网络至基底外侧质膜的运输,突起形成和纤毛发生[28,29]。与对照相比,来自AD脑组织质膜部分的Rab8水平显著增加[30]。另有研究发现,沉默也可增加tau蛋白分泌。Rab1A与高尔基体膜相关,沉默会诱导高尔基体碎裂,这表明Rab介导的高尔基体动力学可能参与调节tau蛋白分泌[31]。Rab3蛋白参与调控APP在轴突的快速运输。沉默和能够降低总体APP蛋白水平,这表明Rab3在APP的运输和维持中发挥了作用[26]。
Ras and Rab Interactor 3 (RIN3)是Rab5 GTPase家族的鸟嘌呤核苷酸交换因子(GEF),美国加州大学圣地亚哥分校医学院的吴承标课题组发现,在AD发病早期,的mRNA水平在皮质和海马中均显著增加,这一现象先于β-淀粉样蛋白沉积产生[32]。在APP/PS1小鼠的基底前脑胆碱能神经元中选择性上调。由于RIN3是Rab5的GEF,因此增加的表达可能有助于APP/PS1小鼠基底前脑胆碱能神经元中Rab5早期内含体的增大。通过质谱分析发现,RIN3向Rab5早期内含体募集了另外两个AD危险因素BIN1和CD2AP,进而损害APP的运输和加工,导致具有神经元毒性的APP-CTF的产生增加。RIN3/BIN1复合物的增加还可以促进Tau蛋白过度磷酸化。Rab5在内含体运输、突触和突触功能中起重要作用[32]。它还参与了兴奋性突触传递的长期增强和抑制过程,这对于学习和记忆功能非常重要[33,34]。Rab5的激活可能会削弱突触功能和细胞之间的通信,导致信号传递到内含体的轴突运输减少和神经元萎缩。
在AD模型鼠中观察到明显增大的溶酶体聚集在一起,显示出溶酶体功能障碍。溶酶体呈现较低的pH值时,Aβ42可能被错误折叠成稳定的聚集体,该聚集体能够在细胞内繁殖并随后成核,并在胞外形成淀粉样斑块[35]。最近发现,参与内含体,自噬体和溶酶体运输的Rab7A可以调节tau蛋白分泌。抑制的表达后,tau蛋白在Thr181和Ser422位点的磷酸化完全被消除,Ser202和Ser404处的磷酸化也显著降低,tau蛋白磷酸化的减少可能有助于其分泌减少。与此一致的是的显性负突变和组成型活性形式的过表达分别减少和增加了tau蛋白分泌[36]。AD患者的人脑和AD小鼠模型脑中可以检测到Rab7A水平升高,且 Rab7A和tau蛋白之间存在共定位现象,这进一步表明Rab7A可能促进tau蛋白在AD中的积累[36]。
4.2 帕金森病
帕金森病也是一种常见的神经退行性疾病,其临床表现为运动缺陷,包括静息性震颤和肌肉僵硬[37]。其病理特征主要是在黑质致密部中积累了由α-突触核蛋白(α-synuclein, α-syn)组成的路易小体和多巴胺能神经元的选择性变性。约95%的PD病例为散发性,其余为家族性。突触核蛋白和的基因突变是引起家族性和散发性帕金森病的主要原因。人类遗传学的最新进展指出,膜运输缺陷是PD致病的关键途径[38]。α-Syn是神经元中高度丰富的蛋白质,溶酶体过程的破坏会影响其代谢[39]。
在PD动物模型中已经证明,过量的α-syn会损害Rab蛋白依赖的细胞内运输,研究发现的过表达可能逆转细胞内毒性,从而阻止神经元的丢失[40]。α-syn的内吞与再循环可能由Rab11A和Rab13介导,同时Rab11A,Rab13和Rab8B可能也参与α-syn在包涵体中的清除[41]。Dinter等[42]发现在HEK293细胞中,的过表达能够增加α-syn的清除率,其效应子FYCO1需要活性Rab7才能发挥作用,继而刺激α-突触核蛋白的降解。在飞行模型中,Rab7和FYCO1可以挽救突变体α-syn诱导的运动功能障碍,因此Rab7可以作为治疗PD的潜在靶点[42]。
LRRK2位于高尔基体、高尔基相关囊泡、内质网、线粒体和溶酶体中,是囊泡运输的一般调节器。LRRK2可能通过调节与内含体分选相关的Rab GTPases (即Rab29、Rab8A、Rab10)在开关II区的保守苏氨酸残基处的磷酸化,进而调节货物的囊泡运输。反过来,LRRK2激酶的活性主要受高尔基复合体内Rab29和内含体中与逆转录子相关的VPS35的调控[43]。磷酸蛋白质组学的结果表明,包括Rab3A、Rab29、Rab8A、Rab10、Rab12和Rab43在内的几种Rab蛋白均是LRRK2的底物[44~46]。通过LRRK2激酶的体外活性测定还发现,Rab GTPase蛋白的一个子集,包括Rab1A、Rab3C、Rab35可以与LRRK2相互作用并被其磷酸化[47]。
越来越多的证据表明,沉默内源性表达导致自噬缺陷,从而使α-syn的异常积累,进而导致PD的发生[39]。Rab29在溶酶体应激条件下,将LRRK2募集到较大的溶酶体中,易位至异常的溶酶体后,LRRK2募集其底物Rab8和Rab10,并通过介导其功能性下游调节子,EH结构域结合蛋白1 (EHBP1)和EHBP1样蛋白1 (EHBP1L1)来促进溶酶体分泌[48]。鉴于EHBP1和EHBP1L1参与内含体和制管膜曲率的形成[49,50],可以推测LRRK2-Rab途径调节溶酶体的形态。
研究证明Rab35是LRRK2的下游效应子。Rab GTPases上的磷酸化位点突变会引起原代皮层神经元中的神经毒性和多巴胺能神经元的变性,这在Rab35磷酸突变体中尤为严重[47]。韩国首尔国立大学医学院李胜在课题组在α-syn转基因小鼠中发现,LRRK2激酶的抑制降低了α-syn的病理特征并增强了α-syn向溶酶体的运输[51]。考虑到Rab35可以通过调节多囊泡体向质膜的运输和对接来调控外泌体的分泌[52],猜测LRRK2-RAB35途径的激活使α-syn避开了溶酶体降解途径,而是导致其进入分泌途径,从而促进了该蛋白的积累[53]。
4.3 肌萎缩性侧索硬化症
肌萎缩性侧索硬化症是一种进行性神经系统变性疾病,主要影响大脑和脊髓中的运动神经元[54]。
与AD和PD相似,所有ALS病例中有90%是散发型的;有10%来自家族,是由编码多种蛋白质的基因突变引起的,这些基因包括(Rab5的鸟嘌呤核苷酸交换因子),(染色体9上的开放读码框72),(超氧化物歧化酶1),(交易反应性DNA结合蛋白43),(肉瘤融合蛋白)和(VAMP (小突触泡蛋白)相关的蛋白B)等[55~57]。
ALS在儿童期发作是由于基因功能丧失的突变所致,ALS2通过激活Rab5在早期内含体中的成熟发挥关键作用。ALS2和Rab5相互作用可以调节神经营养蛋白的信号传导[58]。
在ALS患者的运动神经元中,C9orf72主要定位于Rab5阳性的早期内含体。在神经元细胞系和原代皮层神经元中,C9orf72与Rab1、Rab5、Rab7和Rab11共定位[57]。沉默后,TrkB的内吞作用被抑制,这表明这些患者的内含体运输受到损害[59]。已有报道显示C9orf72与Rab蛋白的相互作用能够调节运动神经元中内含体的运输,该过程是溶酶体发生所必需。C9orf72的耗竭会损害自噬,并导致聚集体的异常聚集,而聚集体是ALS发病的主要特征。沉默后,LC3II:LC3I比例显著增加,表明自噬体形成失调。
Rab1与突变型TDP-43,FUS和SOD1广泛共定位于神经元细胞中,并且Rab1在散发性ALS患者的脊髓运动神经元中参与形成包涵体。SOD1、TDP-43或FUS的突变会导致Rab1的定位出现错误,进而导致内质网–高尔基体网络中的蛋白质运输受损。而这些缺失可以通过的过表达来挽救[60]。的过表达对mSOD1、mTDP-43和mFUS诱导的内质网应激具有保护作用[55]。Rab1介导的内质网–高尔基体运输途径可能是ALS中的新型治疗靶点。
4.4 亨廷顿病
亨廷顿病是一种致命的神经退行性疾病,每100,000名居民中有5~10例患病,典型发病年龄为30~40岁。患者会遭受一系列复杂的精神、认知和运动障碍,直至死亡。亨廷顿病是由亨廷顿基因()中的聚谷氨酰胺()重复扩增引起的常染色体显性遗传病。重复序列的长度与发病年龄成正比,但是遗传变异和环境因素改变了这种相关性[61]。它是由三核苷酸CAG在亨廷顿基因()基因5ʹ末端附近的多态性区域发生异常扩增引起的。这种显性突变不仅耗尽了编码的亨廷顿蛋白,破坏了其生理功能,而且还产生了折叠错误的蛋白,即突变体Htt,并导致纹状体中的中棘神经元死亡[62]。
Rab8 GTPase调节反面高尔基体网管状结构向质膜的运输,optineurin是其效应子,二者形成的复合物在高尔基复合体到基底外侧质膜的极化膜运输过程中以及神经元的树突中起重要作用[63]。的突变与optineurin-Rab8复合物的相互作用减少,从而导致网格蛋白依赖性高尔基体到溶酶体区室的运输发生改变[64]。此外,Htt和HAP40(Htt相关蛋白40)形成的复合物是Rab5的效应子,可控制早期内含体的运动活性[65]。的过表达降低了Htt突变蛋白的聚集,而沉默会抑制内吞作用并阻断自噬,从而增加了polyQ的聚集[66]。Rab11是参与内含体再循环的Rab蛋白,内循环是细胞维持质膜成分的主要途径。突变体Htt抑制Rab11-GDP到Rab11-GTP的核苷酸交换,从而抑制Rab11的活性并导致细胞中转铁蛋白受体的再循环减慢。HD患者中Rab11活性和内体循环功能的异常可能与除转铁蛋白外的许多关键蛋白的转运有关,并对神经元的树突和轴突产生影响[67]。
在HD转基因小鼠中,观察到靠近Htt聚集体的神经元树突棘较少,提示树突棘损失可能是Htt聚集体清除率下降的早期结果。通过在海马神经元中过表达可消除表达Htt的原代鼠神经元中树突棘的丧失,这表明突触功能障碍与Rab11受损可能有早期HD的发病相关[68]。
HD与轴突运输的改变有关。在纹状体神经元中,的表达破坏了轴突的快速运输[69]。在果蝇HD模型中,的过表达能够恢复早期的突触功能障碍,包括突触前囊泡大小的减少,数量振幅的减少和诱发的突触传递以及幼虫爬行的改变等[70]。
4.5 夏科特–玛丽–牙齿2B型
夏科特–玛丽–牙齿(Charcot–Marie-tooth, CMT)病是最常见的遗传性神经肌肉疾病,该病是由于几种不同基因的突变导致相似的表型。CMT主要分为1类(CMT1)和2类(CMT2)形式。CMT1是特征为神经传导速度降低的显性遗传性脱髓鞘神经病,而CMT2是特征为神经传导速度正常或略有降低的显性遗传性轴突神经病。除了这些主要类别外,CMT中还包括其他稀有形式[71]。在此着重介绍直接与Rab相关的CMT疾病,即CMT2B型,其表现包括严重的感觉丧失,远端肌肉无力以及由于反复感染引起的足部溃疡,感染甚至截肢的频繁发生。
Rab7中高度保守的氨基酸残基的5个错义突变与CMT2B型表型相关。点突变引起Rab7蛋白水平升高或降低,进而影响Rab7控制的神经营养蛋白的转运和信号传导、神经突向外生长以及神经元迁移[20,72]。由于神经营养蛋白受体的内吞作用和逆行轴突运输对于神经营养蛋白信号的传导和控制神经元的分化、可塑性和存活至关重要,因此神经营养蛋白运输障碍可能会导致严重的神经变性[73,74]。
在一名CMT2B患者腓肠神经活检中,发现Rab7效应子-Rab相互作用溶酶体蛋白(Rab interacting lysosomal protein, RILP)被下调,导致受体降解和信号衰减。RILP不仅是晚期内体/溶酶体的正常分布所必需的,而且是晚期内体中腔内囊泡的形成所必需的[75,76]。但是,GTP水解不足可能会导致Rab7突变体隔离RILP,从而降低RILP作用于其他方面(如内含体)的能力。由于Rab7是普遍存在的RabGTPase,因此这种罕见疾病也显示了其神经元对膜运输的变化具有敏感性的特点。
5 Rab GTPases介导线粒体和星形胶质细胞的功能
Rab GTPas介导的膜运输与神经退行性疾病的关联是多种多样的。在神经退行性疾病中,在受损线粒体吞噬过程中Rab蛋白同样发挥重要的调控作用。线粒体的状态直接影响神经元的发育、功能和存活。神经元是长寿细胞,在整个生命周期中都存在,因此更容易受到线粒体功能障碍引起的累积损伤的影响。在AD患者神经元内的异常溶酶体中,发现了未消化的受损线粒体[77]。在PD的发病机理中LRRK2、α-Syn会损害线粒体和线粒体功能,因此造成的线粒体受损是必然的。ALS的致病基因涉及线粒体和线粒体调控基因(如、Ser/Thr蛋白激酶()、和超氧化物歧化酶1 ())[78]。功能障碍线粒体的积累已成为患者和动物模型中受累神经元的共同特征,可能出现在明显的认知缺陷之前。
Rab7的活性受两个Rab7 GAPs (TBC1D15和TBC1D17)的调控,同时Rab7也被募集到线粒体外膜,在那里动员囊泡以建立自噬体。TBC1D15和TBC1D17的耗尽会导致Rab7在线粒体上积累,并导致自噬体样结构异常积累,从而延迟并阻碍受损线粒体的清除[79,80]。这些GAPs在空间上控制Rab7活性,Rab7活性本身控制了囊泡向自噬体膜的募集。处于其GTP结合活性状态的Rab32参与线粒体的分裂[81]。Rab35促进自噬受体NDP52的募集以及与泛素的结合,从而促进异种吞噬、线粒体吞噬和自噬体的成熟[82]。
许多研究表明AD、PD和HD等神经退行性疾病的发病机制中存在星形胶质细胞的功能异常,并伴有Rab蛋白水平的变化。神经炎症是所有神经退行性疾病的重要组成部分,其中小胶质细胞和星形胶质细胞通过释放多种促炎和抗炎细胞因子来发挥双向作用[83,84]。小胶质细胞可以通过介导神经炎症调节大脑免疫,并参与突触的连接和重塑;然而在AD早期小胶质细胞可以介导突触的异常丧失进而促进病理进程[85]。星形胶质细胞是大脑中存在较为丰富的细胞,可维持神经递质的稳态,引导突触的形成和成熟,调节活性氧和血脑屏障[86~88]。神经炎症和缺血可诱导出两种不同类型的反应性星形胶质细胞,称为A1和A2反应性星形胶质细胞。A1反应性星形胶质细胞的上调可能是有害的,而A2反应性星形胶质细胞的上调可能是有益的[89]。此外,衰老的星形胶质细胞具有神经炎性A1样反应性星形胶质细胞的反应性表型。除了释放有效的神经毒素外,A1星形胶质细胞还能促进新突触的形成,并导致中枢神经系统神经元的兴奋功能降低。除了星形胶质细胞反应性状态的改变外,星形胶质细胞中可能还会发生其他转录和功能性变化,这可以解释正常衰老过程中认知能力下降这一现象[90],而在PD模型中抑制A1星形胶质细胞的活性具有保护神经作用[91]。
研究发现,在AD中,星形胶质细胞是大脑在生理条件下表达的主要细胞,星形胶质细胞参与Aβ摄取和降解[92,93]。在AD早期阶段,BACE1活性增加伴随着BACE2活性的相应增加,BACE2主要定位于星形胶质细胞中[94]。在PD中,从神经元释放的α-Syn被星形胶质细胞吸收,进而影响其线粒体的完整性并导致神经毒性的产生[95~97]。研究发现星型胶质细胞在神经退行性疾病的病理性扩散中具有潜在的协同作用[98]。在HD中,随着疾病进展会增加相关星形细胞的活性,进而导致Htt的聚集[99,100]。在ALS中,星形胶质细胞显示出毒性表型,引起运动神经元变性。Rab31在表皮生长因子受体转运至晚期内含体的运输中发挥作用[101],沉默研究引起的EGFR信号转导增加可能会阻碍培养物中星形胶质细胞的完全发育,从而导致存活的星形胶质细胞百分比降低[102]。
在促炎条件下,智利大学Quest等[103]通过生物信息学发现,反应性星形胶质细胞中Rab的内吞途径发生了改变,其有利于蛋白水解的回收。特别是,Rab4、Rab5和Rab7的蛋白表达水平发生变化。另一方面,这种促炎环境增加了和的表达,并降低了Rab7-GTP的负荷。在促炎环境中,从早期(Rab5)到晚期内含体(Rab7)的货物运输也发生了变化。通过分析晚期和早期内体组分中的低密度脂蛋白LDL (注定要降解的货物)的分布,研究人员观察到LDL保留在TNFα刺激的细胞的外围,这表明由Rab7所介导的溶酶体降解能力下调[103]。在神经退行性疾病和脑损伤中,星形胶质细胞活化涉及Rab依赖性途径的分子机制尚待研究。
6 结语与展望
本文主要分析了一些Rab蛋白在神经类疾病中的运输和信号传导中的变化,Rab蛋白的异常表达、活性改变或定位错误可能与多种神经类疾病如(AD、PD和HD)的致病机制有关(表1)。参与内吞降解途径的Rab7A可以调节tau蛋白的分泌。Rab11和Rab21可以通过影响BACE1或PS1的定位进而影响Aβ的产生。Rab35可以促进α-syn的分泌。沉默加htt突变蛋白的聚集。在ALS和CMT2B中,Rab7参与调节轴突运输以及内吞途径,神经营养蛋白的信号传导。Rab蛋白主要通过调节内吞途径来影响蛋白聚集体的产生。另一方面,神经退行性疾病的致病机制,即线粒体和星形胶质细胞的功能异常也与Rab蛋白水平的改变密切相关。线粒体功能障碍发生在神经退行性疾病的早期阶段,是造成神经元死亡的重要原因。线粒体功能障碍的出现和加重可能是由病理过程发展中涉及的许多因素造成的。研究与受损线粒体自噬调节的相关Rab蛋白并分析Rab蛋白在其中的调节途径对神经退行性疾病的早期诊断将会有很大帮助。
星形胶质细胞在神经退行性疾病中的作用机制是目前的研究热点,Rab蛋白在其中的作用尚不清楚。星形胶质细胞对于维持脑稳态和保护神经元至关重要。星形胶质细胞中的溶酶体降解能力不足,与包括PD在内的各种神经退行性疾病的发病机理有关。阻止或者减少A1反应性星形胶质细胞的形成可以减缓神经退行性疾病的病理进程。在成年啮齿动物的大脑中,仅在神经胶质纤维酸性蛋白阳性星形胶质细胞中表达[101]。Rab蛋白作为星型胶质细胞的生物标志物,具有作为神经退行性疾病诊断工具的巨大潜力。

表1 神经退行性疾病中的Rab蛋白及其功能
尽管已经非常深入地研究了几种Rab蛋白,但是Rab蛋白家族仍有巨大的研究潜力,Rab蛋白在中枢神经系统不同组织中的特殊作用至关重要。神经退行性疾病是一些常见功能障碍导致的具有不同病理特征的疾病。Rab蛋白在不同的神经退行性疾病中显示出不同的调节作用,但是目前尚不清楚是Rab蛋白的变化引起功能障碍还是功能障碍导致Rab蛋白水平发生变化。发现和研究这些潜在的常见机制可以使我们更加了解神经元存活的基本要求,为深入探究这几种神经类疾病的病理学机制奠定基础,同时为开发有效的治疗策略开辟新的道路。
[1] GBD 2016 Dementia Collaborators. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016., 2019, 18(1): 88–106.
[2] Jia L, Quan M, Fu Y, Zhao T, Li Y, Wei C, Tang Y, Qin Q, Wang F, Qiao Y, Shi S, Wang YJ, Du Y, Zhang J, Zhang J, Luo B, Qu Q, Zhou C, Gauthier S, Jia J. Group for the Project of Dementia Situation in China Dementia in China: epidemiology, clinical management, and research advances., 2020. 19(1): 81–92.
[3] Brennwald P, Novick P. Interactions of three domains distinguishing the Ras-related GTP-binding proteins Ypt1 and Sec4., 1993, 362(6420): 560–563.
[4] Iakovenko A, Rostkova E, Merzlyak E, Hillebrand AM, Thomä NH, Goody RS, Alexandrov K. Semi-synthetic Rab proteins as tools for studying intermolecular interactions., 2000, 468(2): 155–158.
[5] Milburn MV, Tong L, deVos AM, Brünger A, Yamaizumi Z, Nishimura S, Kim SH. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins., 1990, 247(4945): 939–945.
[6] Guadagno NA, Progida C. Rab GTPases: switching to human diseases., 2019, 8(8): 909.
[7] Seabra MC, Wasmeier C. Controlling the location and activation of Rab GTPases., 2004, 16(4): 451–457.
[8] Lin L, Shi AB. Endocytic recycling pathways and the regulatory mechanisms., 2019, 41(6): 451–468.林珑, 史岸冰. 细胞内吞循环运输通路及其分子调控机制. 遗传, 2019, 41(6): 451–468.
[9] Stoorvogel W, Strous GJ, Geuze HJ, Oorschot V, Schwartz AL. Late endosomes derive from early endosomes by maturation, 1991, 65(3): 417–427.
[10] Bucci C, Parton RG, Mather I H, Stunnenberg H, Simons K, Hoflack B, Zerial M. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway, 1992, 70(5): 715–728.
[11] Feng Y, Press B, Wandinger-Ness A. Rab 7: an important regulator of late endocytic membrane traffic, 1995, 131(6 Pt 1): 1435–1452.
[12] Kouranti I, Sachse M, Arouche N, Goud B, Echard A. Rab35regulates an endocytic recycling pathway essential for the terminal steps of cytokinesis., 2006, 16(17): 1719–1725.
[13] Daro E, van der Sluijs P, Galli T, Mellman I. Rab4and cellubrevin define different early endosome populations on the pathway of transferrin receptor recycling., 1996, 93(18): 9559–9564.
[14] Ullrich O, Reinsch S, UrbéS, Zerial M, Parton RG. Rab11 regulates recycling through the pericentriolar recycling endosome, 1996, 135(4): 913– 924.
[15] Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LSB. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in, 2003, 40(1): 25–40.
[16] Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LSB. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease, 2005, 307(5713): 1282–1288.
[17] Dulubova I, Lou Xl, Lu J, Huryeva I, Alam A, Schneggenburger R, Südhof TC, Rizo J. A Munc13/ RIM/Rab3 tripartite complex: from priming to plasticity?, 2005, 24(16): 2839–2850.
[18] Szodorai A, Kuan YH, Hunzelmann S, Engel U, Sakane A, Sasaki T, Takai Y, Kirsch J, Müller U, Beyreuther K, Brady S, Morfini G, Kins S. APP anterograde transport requires Rab3A GTPase activity for assembly of the transport vesicle, 2009, 29(46): 14534– 14544.
[19] Zhang K, Kenan RFB, Osakada Y, Xu W, Sinit RS, Chen L, Zhao XB, Chen JY, Cui BX, Wu CB. Defective axonal transport of Rab7 GTPase results in dysregulated trophic signaling., 2013, 33(17): 7451–7462.
[20] Deinhardt K, Salinas S, Verastegui C, Watson R, Worth D, Hanrahan S, Bucci C, Schiavo G. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway., 2006, 52(2): 293–305.
[21] Binotti B, Pavlos NJ, Riedel D, Wenzel D, Vorbrüggen G, Schalk AM, Kühnel K, Boyken J, Erck C, Martens H, Chua JJE, Jahn R. The GTPase Rab26 links synaptic vesicles to the autophagy pathway., 2015, 4: e05597.
[22] Flament S, Delacourte A, Verny M, Hauw JJ, Javoy- Agid F. Abnormal Tau proteins in progressive supranuclearpalsy. Similarities and differences with the neurofibrillary degeneration of the Alzheimer type, 1991, 81(6): 591–596.
[23] Buggia-Prevot V, Fernandez CG, Riordan S, Vetrivel KS, Roseman J, Waters J, Bindokas VP, Vassar R, Thinakaran G. Axonal BACE1 dynamics and targeting in hippocampal neurons: A role for Rab11 GTPase., 2014, 9: 1.
[24] Udayar V, Buggia-Prévot V, Guerreiro RL, Siegel G, Rambabu N, Soohoo AL, Ponnusamy M, Siegenthaler B, Bali J, Simons M, Ries J, Puthenveedu MA, Hardy J, Thinakaran G, Rajendran L. A paired RNAi and RabGAP overexpression screen identifies Rab11 as a regulator of β-amyloid production, 2013, 5(6): 1536–1551.
[25] Sun ZZ, Xie YJ, Chen YT, Yang QH, Quan ZZ, Dai RJ, Qing H. Rab21, a novel PS1 interactor, regulates γ-secretase activityPS1 subcellular distribution, 2018, 55(5): 3841–3855.
[26] Udayar, V, Buggia-Prévot V, Guerreiro RL, Siegel G, Rambabu N, Soohoo AL, Ponnusamy M, Siegenthaler B, Bali J, Simons M, Ries J, Puthenveedu MA, Hardy J, Thinakaran G, Rajendran L. A paired RNAi and RabGAP overexpression screen identifies Rab11 as a regulator of β-amyloid production, 2013, 5(6): 1536–1551.
[27] Wang X. Pei G. Visualization of alzheimer’s disease related α-/β-/γ-secretase ternary complex by bimolecular fluorescence complementation based fluorescence resonance energy transfer., 2018, 11: 431.
[28] Huber LA, Pimplikar S, Parton RG, Virta H, Zerial M, Simons K. Rab8, a small GTPase involved in vesicular traffic between the TGN and the basolateral plasma membrane, 1993, 123(1): 35–45.
[29] Nachury MV, Loktev AV, Zhang QH, Westlake CJ, Peränen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, Jackson PK. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis,2007, 129(6): 1201– 1213.
[30] Shimohama S, Kamiya S, Taniguchi T, Sumida Y, Fujimoto S. Differential involvement of small G proteins in Alzheimer’s disease, 1999, 3(6): 597–600.
[31] Mohamed NV, Desjardins A, Leclerc N. Tau secretion is correlated to an increase of Golgi dynamics, 2017, 12(5): e0178288.
[32] Shen R, Zhao XB, He L, Ding YB, Xu W, Lin SZ, Fang S, Yang WL, Sung KJ, Spencer B, Rissman RA, Lei M, Ding JQ, Wu CB. Upregulation of RIN3 induces endosomal dysfunction in Alzheimer's disease., 2020, 9(1): 26.
[33] Brown TC, Tran IC, Backos DS, Esteban JA. NMDA receptor-dependent activation of the small GTPase Rab5 drives the removal of synaptic AMPA receptors during hippocampal LTD., 2005, 45(1): 81–94.
[34] Deininger K, Eder M, Kramer ER, Zieglgänsberger W, Dodt HU, Dornmair K, Colicelli J, Klein R. The Rab5 guanylate exchange factor Rin1 regulates endocytosis of the EphA4 receptor in mature excitatory neurons, 2008, 105(34): 12539–12544.
[35] Hu XY, Crick SL, Bu GJ, Frieden C, Pappu RV, Lee JM. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide., 2009, 106(48): 20324–20329.
[36] Rodriguez L, Mohamed NV, Desjardins A, Lippé R, Fon EA, Leclerc N. Rab7A regulates tau secretion, 2017, 141(4): 592–605.
[37] Wissel BD, Dwivedi AK, Merola A, Chin D, Jacob C, Duker AP, Vaughan JE, Lovera L, LaFaver K, Levy A, Lang AE, Morgante F, Nirenberg MJ, Stephen C, Sharma N, Romagnolo A, Lopiano L, Balint B, Yu XX, Bhatia KP, Espay AJ. Functional neurological disorders in Parkinson disease., 2018, 89(6): 566–571.
[38] Hur EM, Jang EH, Jeong GR, Lee BD. LRRK2 and membrane trafficking: nexus of Parkinson’s disease, 2019, 52(9): 533–539.
[39] Schapansky J, Khasnavis S, DeAndrade MP, Nardozzi JD, Falkson SR, Boyd JD, Sanderson JB, Bartels T, Melrose HL, LaVoie MJ. Familial knockin mutation of LRRK2 causes lysosomal dysfunction and accumulation of endogenous insoluble α-synuclein in neurons, 2018, 111: 26–35.
[40] Fleming J, Outeiro TF, Slack M, Lindquist SL, Bulawa CE. detection of compounds that rescue Rab1-synuclein toxicity,, 2008, 439: 339–351.
[41] Gonçalves SA, Macedo D, Raquel H, Simões PD, Giorgini F, Ramalho JS, Barral DC, Moita LF, Outeiro TF. ShRNA-based screen identifies endocytic recycling pathway components that act as genetic modifiers of alpha-synuclein aggregation, secretion and toxicity., 2016, 12(4): e1005995.
[42] Dinter E, Saridaki T, Nippold M, Plum S, Diederichs L, Komnig D, Fensky L, May C, Marcus K, Voigt A, Schulz JB, Falkenburger BH. Rab7 induces clearance of α-synuclein aggregates., 2016, 138(5): 758–774.
[43] Vilariño-Güell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, Behrouz B, Melrose HL, Hentati E, Puschmann A, Evans DM, Conibear E, Wasserman WW, Aasly JO, Burkhard PR, Djaldetti R, Ghika J, Hentati F, Krygowska-Wajs A, Lynch T, Melamed E, Rajput A, Rajput AH, Solida A, Wu RM, Uitti RJ, Wszolek ZK, Vingerhoets F, Farrer MJ. VPS35 mutations in Parkinson disease., 2011, 89(1): 162–167.
[44] Liu ZY, Bryant N, Kumaran R, Beilina A, Abeliovich A, Cookson MR, West AB. LRRK2 phosphorylates membrane-bound Rabs and is activated by GTP-bound Rab7L1 to promote recruitment to the trans-Golgi network, 2018, 27(2): 385–395.
[45] Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S, Lorentzen E, Duddy G, Wilson S, Baptista MA, Fiske BK, Fell MJ, Morrow JA, Reith AD, Alessi DR, Mann M. Phosphoproteomics reveals that Parkinson's disease kinase LRRK2 regulates a subset of Rab GTPases., 2016, 5: e12813.
[46] Steger M, Diez F, Dhekne HS, Lis P, Nirujogi RS, Karayel O, Tonelli F, Martinez TN, Lorentzen E, Pfeffer SR, Alessi DR, Mann M. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis,2017, 6: e31012.
[47] Jeong GR, Jang EH, Bae JR, Jun S, Kang HC, Park CH, Shin JH, Yamamoto Y, Tanaka-Yamamoto K, Dawson VL, Dawson TM, Hur EM, Lee BD. Dysregulated phosphorylation of Rab GTPases by LRRK2 induces neurodegeneration., 2018, 13: 8.
[48] Eguchi T, Kuwahara T, Sakurai M, Komori T, Fujimoto T, Ito G, Yoshimura SI, Harada A, Fukuda M, Koike M, Iwatsubo T. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis., 2018, 115(39): E9115–E9124.
[49] Nakajo A, Yoshimura S, Togawa H, Kunii M, Iwano T, Izumi A, Noguchi Y, Watanabe A, Goto A, Sato T, Harada A. EHBP1L1 coordinates Rab8 and Bin1 to regulate apical-directed transport in polarized epithelial cells, 2016, 212(3): 297–306.
[50] Wang P, Liu H, Wang Y, Liu O, Zhang J, Gleason A, Yang ZR, Wang H, Shi AB, Grant BD. RAB-10 promotes EHBP-1 bridging of filamentous actin and tubular recycling endosomes., 2016, 12: e1006093.
[51] Bae EJ, Kim DK, Kim C, Mante M, Adame A, Rockenstein E, Ulusoy A, Klinkenberg M, Jeong GR, Bae JR, Lee C, Lee HeJ, Lee BD, Monte DAD, Masliah E, Lee SJ. LRRK2 kinase regulates α-synuclein propagationRAB35 phosphorylation., 2018, 9(1): 3465.
[52] Hsu C, Morohashi Y, Yoshimura S, Manrique-Hoyos N, Jung S, Lauterbach MA, Bakhti M, Grønborg M, Möbius W, Rhee J, Barr FA, Simons M. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A–C, 189, 223–232 (2010), 2010.
[53] Bae EJ, Lee SJ. The LRRK2-RAB axis in regulation of vesicle trafficking and α-synuclein propagation, 2020, 1866(3): 165632.
[54] Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, Pagani W, Lodin D, Orozco G, Chinea A. A comprehensive review of amyotrophic lateral sclerosis, 2015, 6: 171.
[55] Soo KY, Halloran M, Sundaramoorthy V, Parakh S, Toth RP, Southam KA, McLean CA, Lock P, King A, Farg MA, Atkin JD. Rab1-dependent ER-Golgi transport dysfunction is a common pathogenic mechanism in SOD1, TDP-43 and FUS-associated ALS., 2015, 130(5): 679–697.
[56] Tsuda H, Han SM, Yang YF, Tong C, Lin YQ, Mohan K, Haueter C, Zoghbi A, Harati Y, Kwan J, Miller MA, Bellen HJ. The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors, 2008, 133(6): 963–977.
[57] Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, Halloran MA, Gleeson PA, Blair IP, Soo KY, King AE, Atkin JD. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking, 2014, 23(13): 3579–3595.
[58] Topp JD, Gray NW, Gerard RD, Horazdovsky BF. Alsin is a Rab5 and Rac1 guanine nucleotide exchange factor, 2004, 279(23): 24612–24623.
[59] Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, Halloran MA, Gleeson PA, Blair IP, Soo KY, King AE, Atkin JD. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking, 2014, 23(13): 3579–3595.
[60] Deshpande M, Feiger Z, Shilton AK, Luo CC, Silverman E, Rodal AA. Role of BMP receptor traffic in synaptic growth defects in an ALS model., 2016, 27(19): 2898–2910.
[61] Djoussé L, Knowlton B, Hayden M, Almqvist EW, Brinkman R, Ross C, Margolis R, Rosenblatt A, Durr A, Dode C, Morrison PJ, Novelletto A, Frontali M, Trent RJA, McCusker E, Gómez-Tortosa E, Mayo D, Jones R, Zanko A, Nance M, Abramson R, Suchowersky O, Paulsen J, Harrison M, Yang Q, Cupples LA, Gusella JF, MacDonald ME, Myers RH. Interaction of normal and expanded CAG repeat sizes influences age at onset of Huntington disease, 2003, 119A(3): 279–282.
[62] Graham SF, Kumar PK, Bjorndahl T, Han B, Yilmaz A, Sherman E, Bahado-Singh RO, Wishart D, Mann D, Green BD. Metabolic signatures of Huntington's disease (HD):1H NMR analysis of the polar metabolome in post-mortem human brain, 2016, 1862(9): 1675–1684.
[63] Sahlender DA, Roberts RC, Arden SD, Spudich G, Taylor MJ, Luzio JP, Kendrick-Jones J, Buss F. Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis, 2005, 169(2): 285–295.
[64] del Toro D, del Toro D, Alberch J, Lázaro-Diéguez F, Martín-Ibáñez R, Xifró X, Egea G, Canals JM. Mutant huntingtin impairs post-Golgi trafficking to lysosomes by delocalizing optineurin/Rab8 complex from the Golgi apparatus, 2009, 20(5): 1478– 1492.
[65] Pal A, Severin F, Lommer B, Shevchenko A, Zerial M. Huntingtin-HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up- regulated in Huntington's disease, 2006, 172(4): 605–618.
[66] Ravikumar B, Imarisio S, Sarkar S, O'Kane CJ, Rubinsztein DC. Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease., 2008, 121(Pt 10): 1649–1660.
[67] Li XY, Standley C, Sapp E, Valencia A, Qin ZH, Kegel KB, Yoder J, Comer-Tierney LA, Esteves M, Chase K, Alexander J, Masso N, Sobin L, Bellve K, Tuft R, Lifshitz L, Fogarty K, Aronin N, DiFiglia M. Mutant huntingtin impairs vesicle formation from recycling endosomes by interfering with Rab11 activity., 2009, 29(22): 6106–6116.
[68] Richards P, Didszun C, Campesan S, Simpson A, Horley B, Young KW, Glynn P, Cain K, Kyriacou CP, Giorgini F, Nicotera P. Dendritic spine loss and neurodegeneration is rescued by Rab11 in models of Huntington’s disease., 2011, 18(2): 191–200.
[69] Her LS, Goldstein LSB. Enhanced sensitivity of striatal neurons to axonal transport defects induced by mutant huntingtin., 2008, 28(50): 13662–13672.
[70] Richards P, Didszun C, Campesan S, Simpson A, Horley B, Young KW, Glynn P, Cain K, Kyriacou CP, Giorgini F, Nicotera P. Dendritic spine loss and neurodegeneration is rescued by Rab11 in models of Huntington's disease., 2011, 18(2): 191–200.
[71] De Luca A, Progida C, Spinosa MR, Alifano P, Bucci C. Characterization of the Rab7K157N mutant protein associated with Charcot–Marie-Tooth type 2B., 2008, 372(2): 283–287.
[72] Saxena S, Bucci C, Weis J, Kruttgen A. The small GTPase Rab7 controls the endosomal trafficking and neuritogenic signaling of the nerve growth factor receptor TrkA., 2005, 25(47): 10930–10940.
[73] Bronfman, FC, Escudero CA, Weis J, Kruttgen A. Endosomal transport of neurotrophins: roles in signaling and neurodegenerative diseases., 2007, 67(9): 1183–1203.
[74] Moises T, Dreier A, Flohr S, Esser M, Brauers E, Reiss K, Merken D, Weis J, Krüttgen A. Tracking TrkA’s trafficking: NGF receptor trafficking controls NGF receptor signaling., 2007, 35(2): 151– 159.
[75] Progida C, Cogli L, Piro F, De Luca A, Bakke O, Bucci C. Rab7b controls trafficking from endosomes to the TGN, 2010. 123(9): 1480.
[76] Progida C, Malerød L, Stuffers S, Brech A, Bucci C, Stenmark H. RILP is required for the proper morphology and function of late endosomes, 2007, 120(21): 3729–3737.
[77] Ye X, Sun XQ, Starovoytov V, Cai Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer's disease patient brains, 2015, 24(10): 2938–2951.
[78] Moore AS, Holzbaur ELF. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy, 2016, 113(24): E3349–E3358.
[79] Yamano K, Fogel AI, Wang CX, van der Bliek AM, Youle RJ. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy., 2014, 3: e01612.
[80] Yamano K, Wang CX, Sarraf SA, Münch C, Kikuchi R, Noda NN, Hizukuri Y, Kanemaki MT, Harper W, Tanaka K, Matsuda N, Youle RJ. Endosomal Rab cycles regulate Parkin-mediated mitophagy, 2018, 7: e31326.
[81] Alto NM, Soderling J, Scott JD. Rab32 is an A-kinase anchoring protein and participates in mitochondrial dynamics., 2002, 158(4): 659–668.
[82] Minowa-Nozawa A, Nozawa T, Okamoto-Furuta K, Kohda H, Nakagawa I. Rab35 GTPase recruits NDP52 to autophagy targets, 2017, 36(18): 2790–2807.
[83] Minkiewicz J, de Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome., 2013, 61(7): 1113–1121.
[84] Koenigsknecht J, Landreth G. Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin- dependent mechanism., 2004, 24(44): 9838– 9846.
[85] Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li SM, Ramakrishnan S, Merry KM, Shi QQ, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B. Complement and microglia mediate early synapse loss in Alzheimer mouse models.,, 2016, 352(6286): 712–716.
[86] Diniz LP, Almeida JC, Tortelli V, Vargas Lopes C, Setti-Perdigão P, Stipursky J, Kahn SA, Romão LF, de Miranda J, Alves-Leon SV, de Souza JM, Castro NG, Panizzutti R, Gomes FC. Astrocyte-induced synaptogenesis is mediated by transforming growth factor β signaling through modulation of D-serine levels in cerebral cortex neurons, 2012, 287(49): 41432–41445.
[87] Drukarch B, Schepens E, Stoof JC, Langeveld CH, Van Muiswinkel FL. Astrocyte-enhanced neuronal survival is mediated by scavenging of extracellular reactive oxygen species., 1998, 25(2): 217–220.
[88] Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions, 2004, 431(7005): 195–199.
[89] Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential., 2017, 46(6): 957–967.
[90] Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity, 2018, 115(8): E1896–E1905.
[91] Yun SP, Kam T, Panicker N, Kim S, Oh Y, Park J, Kwon S, Park YJ, Karuppagounder SS, Park H, Kim S, Oh N, Kim NA, Lee S, Brahmachari S, Mao XB, Lee JH, Kumar M, An D, Kang S, Lee Y, Lee KC, Na DH, Kim D, Lee SH, Roschke VV, Liddelow SA, Mari Z, Barres BA, Dawson VL, Lee S, Dawson TM, Ko HS. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease., 2018, 24(7): 931–938.
[92] Koistinaho M, Lin SZ, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides, 2004, 10(7): 719–726.
[93] Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, Cam HP, Gjoneska E, Raja WK, Cheng J, Rueda R, Kritskiy O, Abdurrob F, Peng ZY, Milo B, Yu CJ, Elmsaouri S, Dey D, Ko T, Yankner BA, Tsai LH. APOE4 causes widespread molecular and cellular alterations associated with alzheimer’s disease phenotypes in human iPSC-derived brain cell types., 2018, 98(6): 1141–1154.e7.
[94] Holler CJ, Webb RL, Laux AL, Beckett TL, Niedowicz DM, Ahmed RR, Liu YX, Simmons CR, Dowling ALS, Spinelli A, Khurgel M, Estus S, Head E, Hersh LB, Murphy MP. BACE2 expression increases in human neurodegenerative disease, 2012, 180(1): 337–350.
[95] Lindström V, Gustafsson G, Sanders LH, Howlett EH, Sigvardson J, Kasrayan A, Ingelsson M, Bergström J, Erlandsson A. Extensive uptake of α-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage, 2017, 82: 143–156.
[96] Braidy N, Gai WP, Xu YH, Sachdev P, Guillemin GJ, Jiang XM, Ballard JWO, Horan MP, Fang ZM, Chong BH, Chan DKY. Uptake and mitochondrial dysfunction of alpha-synuclein in human astrocytes, cortical neurons and fibroblasts, 2013, 2(1): 20.
[97] Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E, Lee SJ. Direct transfer of α-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies, 2010, 285(12): 9262–9272.
[98] Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, Singh I, Deane R, Nedergaard M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury, 2014, 34(49): 16180–16193.
[99] Singhrao SK, Thomas P, Wood JD, MacMillan JC, Neal JW, Harper PS, Jones AL. Huntingtin protein colocalizes with lesions of neurodegenerative diseases: An investigation in Huntington's, Alzheimer's, and Pick's diseases, 1998, 150(2): 213–222.
[100] Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity, 2005, 171(6): 1001–1012.
[101] Ng EL, Ng JJ, Liang F, Tang BL. Rab22B is expressed in the CNS astroglia lineage and plays a role in epidermal growth factor receptor trafficking in A431 cells., 2009, 221(3): 716–728.
[102] Chua CEL, Goh ELK, Tang BL. Rab31 is expressed in neural progenitor cells and plays a role in their differentiation., 2014, 588(17): 3186-3194.
[103] Díaz J, Quest A, Leyton L. Increased expression of αvβ3 integrin in reactive astrocytes is controlled by the Rab endocytic pathway., 2019, 29: S462–S463.
The roles of Rab protein family in neurological diseases
Anping Wu, Hong Qing, Zhenzhen Quan
,,,
The intracellular membrane trafficking is a complicated pathway network. Rab GTPases are key regulators of membrane trafficking that are generally considered as specific markers and indicators of various organelles and membrane trafficking in endocytic and secretory pathways. Dysfunction in axonal and endosomal transport related to Rab proteins is one of the most important causes of neurodegenerative diseases. In this review, we mainly introduce how the Rab proteins change in different neurodegenerative diseases and their regulatory roles in the pathological mechanisms of related diseases. We also discuss the relationships between mitochondrial and glial cell dysfunctions and Rab proteins. Further exploration of the regulatory roles of Rab proteins will shed lights on revealing the pathogenic mechanisms of neurological diseases and providing potential targets for the early diagnosis and treatment of neurological diseases.
Rab proteins; neurological diseases; membrane trafficking; mitochondria; astrocytes
2020-11-17;
2021-01-06
国家自然科学基金项目(编号:81701260)资助[Supported by the National Natural Science Foundation of China (No. 81701260)]
吴安平,在读硕士研究生,专业方向:生物工程。E-mail: 18801361945@163.com
全贞贞,博士,副研究员,研究方向:阿尔茨海默病的分子机制。E-mail: qzzbit2015@bit.edu.cn
10.16288/j.yczz.20-318
2021/1/13 13:37:12
URI: https://kns.cnki.net/kcms/detail/11.1913.R.20210112.1032.001.html
(责任编委: 史岸冰)
猜你喜欢
传染病信息(2022年2期)2022-07-15
生物化工(2021年2期)2021-01-19
生物化工(2020年1期)2020-02-17
读与写(2019年35期)2019-11-05
数学物理学报(2019年3期)2019-07-23
现代职业教育·高职高专(2018年7期)2018-05-14
老年医学与保健(2017年6期)2017-02-06
中华肩肘外科电子杂志(2017年1期)2017-01-11
中外医疗(2015年5期)2016-01-04
中国中医药现代远程教育(2014年13期)2014-03-01
