
可变剪接在神经精神疾病中的作用研究进展
2021-01-18 13:12:12黄鸣鹤吴海涛
中国药理学与毒理学杂志 2020年10期
黄鸣鹤,吴海涛,2
(1.南华大学衡阳医学院,湖南 衡阳 421001;2.军事科学院军事医学研究院军事认知与脑科学研究所,北京 100850)
真核生物基因表达调控可在多个层面进行,包括基因水平、转录和转录后水平、翻译和翻译后水平,其中编码基因DNA中包含外显子和内含子,在转录生成前体mRNA后,在剪接复合物作用下,可对外显子和内含子进行拼接,产生不同的成熟mRNA转录本,此过程谓之可变剪接(alternative splicing,AS)。此后,不同mRNA转录本在核糖体内翻译合成各种蛋白质,行使各种复杂的生物学功能。
AS于1977年由ROBERTS和SHARP实验室首次发现并报道。次年,GALLEGO-PAEZ[1]建议将成熟mRNA中包含和剔除的片段分别命名为“外显子”和“内含子”。AS是基因表达过程中的普遍调节机制,其可从单个基因中产生1个以上的独特mRNA种类。AS可产生在非翻译区(untranslated region,UTR)或编码序列中不同的mRNA,其机制包括外显子跳跃、互斥外显子之间的选择、替代剪接位点的使用和内含子保留[2]。这些差异可能会影响mRNA的稳定性、定位或翻译[3]等功能。高通量研究发现,>95%人类多外显子基因存在AS[4]。
近年来,神经系统中的RNA剪接调控一直是领域内的重要研究方向之一[5]。随着对脊椎动物神经系统发育和神经精神疾病研究的逐步深入,越来越多研究表明,AS在神经元轴突生长、生长锥引导、突触发生和离子通道活动等过程中发挥关键作用[6]。一些研究也提示,神经元发育和稳态维持对于AS扰动更为敏感,AS在多种发育性神经精神疾病进程中发挥着关键作用[7]。
1 可变剪接的基本生物学过程
AS是一种进化上保守的转录后过程,它增加了真核生物中RNA和蛋白质的多样性。不仅大多数基因可编码AS的前体mRNA,而且单个基因编码的mRNA亚型数量可从2个到几千个不等,如果蝇基因唐氏综合征细胞黏附分子,它可产生38 016个不同的mRNA异构体[8]。AS可作为基因表达的开关,进而影响mRNA的表达水平、稳定性、半衰期和定位,形成生物学功能和特性的多样性。
剪接过程主要由剪接体催化介导,其中起到关键作用的剪接体由5个小核核糖核蛋白(small nuclear ribonucleoprotein,snRNP)微粒U1,U2,U4,U5和U6和许多蛋白质因子组成的复合体。snRNP以顺序方式与前体mRNA结合,在U4/U6.U5-snRNP结合后,剪接体经历了长时间的结构重塑过程,导致U1和U4的释放,19复合物相关蛋白(nineteen complex-related proteins)的增加和剪接体的激活。催化剪接体经历两次连续的酯交换反应,从而连接外显子并释放内含子套索[9]。剪接反应完成后,催化后剪接体解离,释放成熟的mRNA,然后与19复合物相关蛋白结合,并在新的剪接循环中分解所有组分[10]。
真核生物转录生成的RNA分子是前体RNA,也称为初级RNA转录产物。几乎所有初级RNA转录产物都需要经过加工才能成为功能性的成熟RNA,加工主要在细胞核内进行。真核生物前体mRNA合成后,需要进行5′和3′端修饰,对前体mRNA进行剪接后才能成为成熟mRNA,再被转运到核糖体指导蛋白质翻译。AS是一种剪接改变机制。前体mRNA的外显子被AS以不同顺序连接形成具有不同蛋白质编码序列和RNA调节元件的大量转录本[11]。目前认为,存在7种AS的通用模型,包括盒式外显子、互斥外显子、竞争5′或3′剪接位点、内含子保留、选择性启动子和可变3′多聚腺苷化位点(图1)。研究显示,在组织特化和发育阶段,>90%的人类基因在经历AS处理后发生明显改变[12]。AS很少对刺激产生“是或否”的反应,通常多个AS产物共存于单个细胞中,使细胞有能力处理不同的内部和外部刺激。有趣的是,多达1/3的AS转录本会产生提前终止密码子,并通过无义突变介导RNA的衰变。在神经系统中,几千个AS事件在离子运输、受体识别、神经传递和学习记忆中发挥重要调控作用[13]。相对而言,AS的不当调控也可能导致使某些神经系统相关疾病发生。
2 可变剪接与神经发育性疾病
在整个脊椎动物进化过程中,AS程序在神经系统中最为复杂,其可能与大脑复杂的解剖结构、发育和功能密切相关。相较于其他组织,大脑具有更多的AS事件发生。另一方面,其对大脑的保护也至关重要,大脑皮质作为认知信息加工处理的核心也受到AS程序的严格调控。大脑发育过程中,大多数AS事件多发生在神经元及其前体细胞内,而非胶质细胞、上皮细胞或其他非神经细胞[14]。因此,AS可精准动态调控是大脑皮质细胞群体形成的必要条件,而参与AS调节的神经RNA结合蛋白(RNA-binding protein,RBP)的突变和神经AS模式的异常也被证明与神经系统疾病有关。换言之,AS调控的异常可导致神经发育性疾病产生。
2.1 小头症
小头症是一类脑部发育不全性神经疾病,其成因与神经发育早期神经前体细胞的增殖、迁移和分化障碍密切相关。最近有临床研究发现,一个编码剪接体U5蛋白质的延长因子Tu GTP蛋白结合域 2(elongation factor Tu GTP binding domain containing 2,Eftud2)基因突变可导致严重的伴有小头症的中颌面骨不全(mandibulofacial dysostosis with microcephaly,MFDM)。MFDM患者的特点是颧骨和下颌发育不全、小头畸形、外耳畸形和智力障碍[15]。Eftud2与核糖体翻译延伸因子EF-2非常相似。它是一个高度保守的剪接体GTPase蛋白家族。U5 snRNP是构象重塑中的主要和次要剪接小体机制所必需的,这是剪接小体激活的关键阶段。Eftud2是U5 snRNP的核心成分,在酿酒酵母中破坏Eftud2的GTP结合域可导致mRNA剪接不全和致死性,表明Eftud2在RNA剪接中具有不可替代的作用[16]。
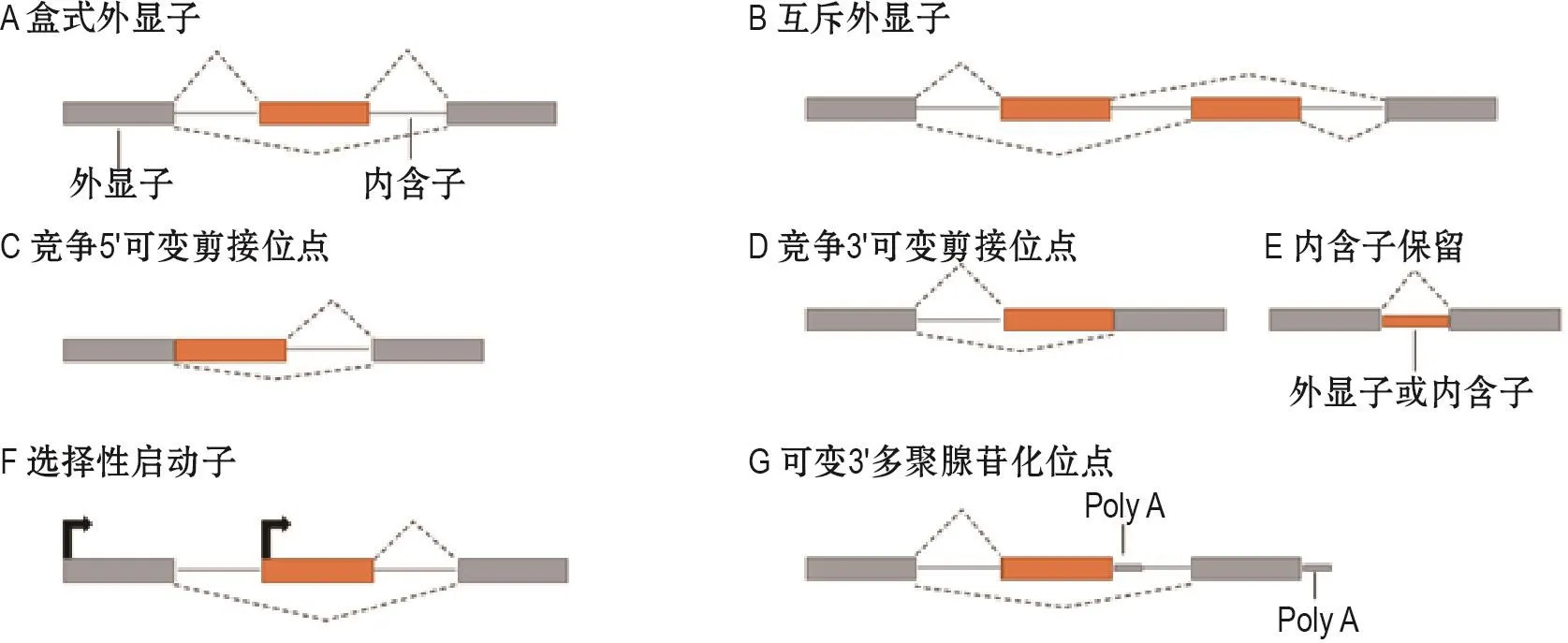
图1 可变剪接的常见模式.A:盒式外显子选择性剪接,包括或排除mRNA中的整个外显子;B:互斥外显子是指1对连续的外显子,其中只有1个包含在内;C:竞争的5′剪接是在1个外显子内(通常是2个)连续的5′剪接位点竞争;D:竞争的3′剪接通过连续的3′剪接位点之间的竞争;E:内含子保留是指1个典型的切除内含子保留在最终的mRNA中;F:选择启动子是指mRNA的不同第1外显子;G:可变3′多聚腺苷化位点是指不同的最后外显子,因此具有不同的3′UTR.
有研究利用全外显子组测序技术,在一例非综合征性原发性小头畸形和智力障碍患者中,发现SnRNP多肽E基因存在一个新的杂合子错义突变,这种突变产生的蛋白产物不能与SMN复合物结合,故不能组装成剪接体U snRNP[17]。人类多嘧啶束结合蛋白1(polypyrimidine tract binding protein 1,PTBP1)结合位点一个内含子突变,就能扰乱神经前体细胞内细丝蛋白A外显子的正常跳跃,从而导致脑特异性畸形[18]。研究还发现,外显子连接复合体因子外显子连接复合物亚基的单倍缺失与人类小头畸形表型密切相关[19],机制上可能与大脑外显子拼接跳跃有关。
2.2 自闭症谱系障碍(autistic spectrum disorder,ASD)
ASD的神经生物学特征之一是突触发育异常和神经环路兴奋/抑制失衡,表现为神经网络整体功能的紊乱[20]。自闭症大脑转录图谱显示,在超过1/3的被分析个体中,神经元剪接调节因子丝氨酸/精氨酸重复矩阵4(serine/arginine repetitive matrix 4,SRRM4)及其目标微外显子剪接程序存在错误调节。有报道通过培育相关蛋白及靶剪接水平降低的突变小鼠,证明相当一部分自闭症患者同nSR100依赖性剪接网络调控障碍有关[21]。
一项从ASD男患儿血液样本中分离出的RNA转录表达研究发现,同其他正常发育男童相比,ASD患儿体内发现了53个差异剪接基因[22]。此外,AS以及其他形式的RNA加工和输运都是响应神经元活性变化而动态调节的[23]。通过利用微阵列和高通量RNA测序技术对自闭症大脑进行转录组图谱分析,已部分揭示了错误调控基因表达和AS的常见模式[24]。在ASD患者脑组织中发现,RNA结合剪接调节因子Rbfox家族的转录水平降低,这些组织也具有5%~10%的替代外显子错误调控[25]。RNA结合fox-1同源物1(RNA binding fox-1 homolog 1,Rbfox1)被证明可调节选择性剪接、mRNA稳定性以及与脑发育和ASD相关基因的翻译[26],凸显了Rbfox目标网络在自闭症中潜在的临床意义。
2.3 Rett综合征(Rett′s syndrome,RTT)
RTT是一种严重的出生后神经发育障碍性精神疾病,其致病基因是X连锁基因甲基-CpG结合蛋白2(methyl-CpG binding protein 2,MECP2)的突变或缺失[27]。其特征是出生后12~18月龄都可正常发育,却在语言和运动技能上退化,并伴有其他神经精神症状和行为异常,如癫痫发作、学习障碍和自闭症等[28]。研究表明,MECP2可调节转录和染色质构象,控制微RNA加工并调节RNA剪接。MECP2与剪接因子相互作用可调节谷氨酸受体基因的剪接,介导了大脑中绝大多数兴奋性突触传递。基于MECP2与剪接因子之间的直接相互作用,基本确定了RTT小鼠模型中特定RNA剪接变化可能与突触之间异常功能连接表型密切相关[29]。
2.4 运动神经元病
脊髓性肌萎缩症(spinal muscular atrophy,SMA)是一种罕见的常染色体隐性遗传病,由脊髓和脑干的运动神经元退化引起。每6000~10 000个活产婴儿中就有1例发生SMA[30],SMA是由存活运动神经元1(survival of motor neuron 1,SMN1)基因的纯合子缺失或突变导致脊髓和脑干中SMN蛋白表达降低和运动神经元变性引起的[31],而其同样编码SMN蛋白的同源基因SMN2由于异常剪接,90%~95%翻译蛋白被截短且无功能,从其产生的功能全长SMN蛋白水平来看SMN2仅为SMN1产生水平的5%~10%,因为SMN2中的剪接位点变体导致从成熟RNA转录物中排除第7外显子,并产生截短、功能失调的SMN蛋白[32],因此,调节SMN2的前体mRNA剪接以促进SMN蛋白的产生可能在SMA的疾病谱中是一种有效的治疗策略。
值得一提的是,2016年美国食品和药物管理局批准上市了一种治疗SMA的反义寡核苷酸药物nusinersen,该药物可修饰SMN2前体mRNA剪接,以促进SMN蛋白的增加。在婴儿期SMA中开展的Ⅱ期开放标记剂量研究表明,接受该药治疗的患儿运动功能逐渐改善,生存期显著延长[33]。
3 可变剪接与精神类疾病
AS在复杂的遗传性疾病,如神经发育和精神障碍中的作用尚未被充分了解。不过,在临床精神分裂症、重度抑郁障碍(major depressive disorder,MDD)和双相情感障碍(bipolar disorder,BP)患者的组织中的确发现了RNA剪接异常的存在。研究表明,存在与转录异构体数量变化密切相关的遗传变异,这些基因变异富含与精神疾病相关的位点,以及根据反式剪接作用因子的改变而导致剪接网络变化[34]。
3.1 重度抑郁障碍
MDD是一种以情绪低落、动机下降、快感和兴趣感丧失为特征的常为慢性且十分严重的精神障碍[35]。5-羟色胺1A(5-hydroxytryptamine 1A,5-HT1A)受体是5-HT能活性的关键调节因子,与情感和情绪有关。人类5-HT1A受体的RNA通过剪接去除mRNA位点产生超稳定的RNA来增加5-HT1A受体的表达。而在重度抑郁症患者中这种剪接明显减少。
在小鼠模型上,突触前和突触后5-HT1A受体的表达决定了抑郁和焦虑样行为,以及对抗抑郁药物的反应,抑制5-HT1A自身受体增加5-HT神经元活性,增强应激恢复力/抗抑郁反应,而其过表达则有相反的作用,海马中的突触后5-HT1A异源受体对抗抑郁药反应至关重要,而前额叶皮质中的异源受体保护免受焦虑和抑郁表型的影响[36]。在人类中,尤其是在PFC中,突触后5-HT1A受体减少与抑郁和焦虑密切相关。LE FRANCOIS等[37]认为这种新型剪接事件是由PTBP1和SRRM4拮抗的。同样,抑郁个体的PFC中5-HT1A受体的RNA剪接率降低,也与SRRM4表达降低有关。这些结果都提示,AS和剪接因子会在大脑基因表达调控中发挥重要功能。
3.2 双相情感障碍
研究表明,BP患者大脑皮质和海马不同区域的糖皮质激素受体(glucocorticoid receptor,GR)表达均显著减少[38],GR有几种通过选择性剪接产生的亚型,其中2种(GRα和GRβ)因其相对丰度较高而成为大多数研究的重点[39],尽管健康对照组的GRα和GRβ mRNA表达水平呈显著负相关,但无论是抑郁症还是BP患者都未有研究显示这种相关性。这些发现提示在情绪障碍患者中可能存在选择性剪接缺陷机制[40]。
最近有研究已确定了一些BP相关危险基因的剪接变体,如全基因组关联研究(genome-wide association studies,GWAS)对BP的研究已经确定了锚蛋白3(ankyrin 3,ANK3)基因中相关的遗传变异。研究发现,在ANK3基因一个含有54个核苷酸的小外显子剪接位点上有一个变体的次要等位基因造成了一个功能丧失的位点,并且这个外显子没有包括在ANK3的转录本中[29]。因此,这种微小的等位基因和外显子跳跃事件被认为对BP和精神分裂症有保护作用。
3.3 精神分裂症
精神分裂症患者通常表现出包括妄想、幻觉、无组织的言语和行为等精神及认知功能障碍。一项研究使用RNA测序技术对9名患者和9名对照的颞上回组织进行了分析,确定了1000多名精神分裂症患者与对照相比存在显著差异剪接的基因[41]。通过对20名精神分裂症患者和20名匹配者尸检背外侧前额叶组织进行RNA测序分析,鉴定出316个基因中798个差异调控转录本,这些转录本富含参与炎症反应的基因。基于在一个苏格兰大家族中发现的染色体易位研究,精神分裂症敏感基因被确定为精神障碍的潜在易感基因,并且已被证明产生50多个转录体亚型,其中一些水平似乎与风险基因型相关。与对照组相比,精神分裂症患者海马中缺失外显子7和8的转录本以及具有差异表达外显子的转录本表达更高[42]。并且精神分裂症敏感基因位点外显子7和8缺失转录本,以及外显子3缺失转录本均与精神分裂症显著相关。
锌指蛋白804A(zinc finger protein 804A,ZNF804A)编码一个假定的锌指转录因子,该转录因子首先由GWAS鉴定为精神分裂症和BP相关基因[43]。体内存在不同程度地表达一个缺少外显子1和2的ZNF804A转录本,该转录本被认为会移除假定的锌指结构域。同对照组相比,在精神分裂症患者中,这一外显子1和2跳过转录本的表达水平较低,在风险基因型纯合子BP患者中表达水平也较低。与之相反,同样的转录本在MDD患者中的表达水平更高[44],提示ZNF804A的不同剪接方式可能会影响神经网络和神经递质传递的功能。
多巴胺受体2(dopamine receptor 2,D2)基因一直被认为是形成精神疾病的风险基因,GWAS研究已经在D2基因附近发现了与精神分裂症密切相关的重要标记。最近发现位于内含子6的D2变异与外显子6选择性剪接和中国汉族人群精神分裂症风险有关[45]。体外寡核苷酸分析表明,D2变异体与另一种剪接调节因子ZRANB2的结合亲和力降低。进一步研究表明,该变异还与短长D2异构体的比率降低有关。
3.4 成瘾、睡眠障碍和其他
Fragile X精神发育迟滞蛋白(Fragile X mental retardation protein,FMRP)是一种多核糖体相关的神经元mRNA结合蛋白,其靶向和翻译抑制与突触可塑性相关的mRNA,并与自闭症、情感障碍、多动症、双相情感障碍、精神分裂症和成瘾有关[46]。FMRP靶向数百种参与细胞骨架重塑的神经发育和神经可塑性蛋白质,其中许多靶点与成瘾有关,包括代谢型谷氨酸受体 1(metabotropic glutamate receptor 1,mGluR1)、mGluR5、大的MAGUK支架蛋白4、细胞质FMR1相互作用蛋白1等[47]。
异质核糖核蛋白(heterogeneous ribonucleoprotein,hnRNP)是一大类多样性核浆定位多功能RNA结合蛋白,它们通过形成RNP复合物,参与调节mRNA的剪接、输出、定位、翻译和稳定性。越来越多的证据表明,hnRNP如hnRNP H1可能参与多种成瘾[48]。哺乳动物睡眠相关昼夜节律与AS的关系源自基于微阵列研究的发现,研究表明,大约有62种RNA结合蛋白可能与睡眠相关RNA加工事件的调控有关。值得一提的是,可变剪接因子类似4的U2小核RNA辅助因子1(U2 small nuclear RNA auxiliary factor 1 like 4,U2AF1L4)基因敲低小鼠在睡眠调节方面存在显著缺陷[49]。
最新研究还表明,突触形成前细胞黏附因子Ⅱ型受体PTPδ的整体缺失会破坏兴奋性突触的形成和非快速眼球转动睡眠。PTPδ条件性敲除或PTPδ突变小鼠在睡眠行为和节律方面均出现显著异常,表明AS在调节兴奋性突触发育、睡眠行为和节律中均发挥重要作用[50]。
4 可变剪接与神经退行性疾病
从全球范围看,包括阿尔茨海默病(Alzheimer disease,AD)、帕金森病(Parkinson disease,PD)和亨廷顿病(Huntington Disease,HD)等在内的神经退行性疾病发生率呈逐年增加的态势,越发受到国际关注。目前,虽然对神经退行性疾病的常见致病机制(如蛋白质聚集或功能障碍、免疫反应改变和轴突变性等)已有基本了解,但在RNA剪接层面的病理生理学调控机制仍不明了。因此,了解神经特异性选择性剪接如何影响大脑基因转录调控进程,对揭示神经退行性疾病的病理生理学机制具有重要意义。
4.1 阿尔茨海默病
AD是临床上最常见的神经退行性疾病[51],AD的神经病理学特征在于细胞外β淀粉样蛋白沉积,以及高磷酸化tau蛋白的积累导致神经原纤维缠结[52]。研究表明,AS参与了AD患者脑内功能变化相关的基因表达异常调控进程。最新针对AD患者脑内蛋白质组学特征的研究,揭示了不溶性U1 snRNP聚集增加,提示核心剪接机制可能在AD发生过程中产生变化[53]。RNA结合蛋白作为一类在AD中增加的蛋白家族出现,并且这些蛋白在与tau缠结病理相关的模块中富集。通过对AD患者、无症状AD和对照人群死后大脑的加权蛋白质相关网络分析表明,因RNA结合蛋白功能障碍导致的RNA剪接变化可能在AD病理进程中发挥重要作用[54]。
JOHNSON等[54]开发了一条新的蛋白质组学管道,用于预测AD患者脑内常规蛋白质数据库中无法检测到的外显子-外显子连接剪接事件。结果发现,许多已确定的替代性外显子-外显子连接剪接事件可能与AD发生密切相关,提示RNA异常剪接在AD发病进程中发挥潜在作用。研究还表明,AD患者脑内背外侧前额叶皮质中磷脂酰肌醇结合网格蛋白组装蛋白,丛生蛋白和蛋白酪氨酸激酶2 β等基因可发生剪接改变。进一步通过转录组范围内关联研究,确定了与AD密切关联的21个基因[55]。相关研究进一步表明,神经元内mRNA异常剪接机制与AD发生密切相关。
4.2 帕金森病
PD由蛋白质异常聚集引起,如路易小体,进而引发中脑黑质多巴胺能神经元丢失[56]。多巴胺缺乏、蛋白酶体功能障碍、线粒体复合物-1活性下降、异常的可变剪接和氧化应激是PD的特征表现[57]。SHEHADEH等[58]对PD患者血液进行基因芯片研究发现,编码丝氨酸/精氨酸重复矩阵2(serine/arginine repetitive matrix 2,SRRM2)的AS相关基因是所有3种PD分析中唯一差异上调的基因,并且在PD患者血液中发现218个基因存在显著差异的外显子表达现象。SRRM2包含2种主要AS变异体,分别为长亚型SRRM2和缺少外显子12~外显子15的短亚型SRRM2转录本,PD患者脑内杏仁核和黑质中均表现为长亚型SRRM2转录本低表达,而短亚型SRRM2转录本高表达。通过对17例PD患者外周血进行外显子微阵列分析发现,SRRM2上游外显子显著上调,而下游外显子下调,最终导致长亚型异构体表达降低。
富含亮氨酸重复激酶2(leucine rich repeat kinase 2,LRRK2)异常过度表达可导致2个PD相关基因剪接改变,即微管相关蛋白tau第10外显子和α-突触核蛋白(α-synuclein,SNCA)基因第5外显子增加。ELLIOTT[59]团队基于过表达野生型LRRK2或其G2019S突变体的细胞外显子阵列分析研究表明,LRRK2可显著影响AS过程,其G2019S突变体与神经退行性基因剪接改变密切相关。SNCA相互作用蛋白基因编码的Synfilin-1是一种突触前结合蛋白,E3泛素蛋白连接酶Parkin,Synfilin-1和SNCA共表达可促进类似于路易小体的细胞质内含物形成。而有研究证明PD患者脑内的确存在缺少外显子3和4,含有外显子9A的Synfilin-1A,Synfilin-1A的过度表达导致蛋白酶体饱和,并直接促进蛋白质包含、形成和神经毒性[10]。此外,2个与神经退行性变密切相关的突变基因——TAR DNA结合蛋白(TAR DNA binding protein,TARDBP)和FUS RNA结合蛋白(FUS RNA binding protein,FUS)基因均编码hnRNP,进一步表明,神经退行性疾病与外显子剪接和转录调控密切相关[59]。
4.3 亨廷顿病
HD是一种遗传性神经退行性疾病[60],其症状包括进行性运动功能障碍、舞蹈症、认知障碍和精神异常,并最终导致患者死亡[61]。该病系基因突变引起,该突变使亨廷顿蛋白(Huntingtin,HTT)基因外显子1中的聚谷氨酰胺CAG三核苷酸重复序列扩增。突变的HTT蛋白可破坏自噬和正常的囊泡转运,导致神经递质信号转导和线粒体功能紊乱[62]。在HD中,RNA的致病机制主要包括选择性剪接的干扰、异常基因沉默、转录产物的异常亚细胞定位和核仁应激等[63]。目前已知隔离剪接因子6介导的2种剪接改变可能与HD发生密切相关,其中一个在HTT基因本身[64],另一个是HD患者的神经变性导致tau基因第10外显子错配[65]。
HD还与微管相关蛋白2(microtubule associated protein 2,MAP2)和tau的异常剪接密切相关。观察到HD患者MAP2总体水平降低及在亚细胞定位上的异常重新分布,从而导致HD神经元树突萎缩。此外,可偶发核凹陷的MAP2阳性结构,并与tau共定位。因此,MAP2/tau含量失衡以及MAP2功能改变可能与HD纹状体萎缩和功能障碍有关。有研究表明,可以通过利用剪接体介导的反式剪接来干预和修复突变型HTT,并开展了RNA治疗策略的优化研究[66]。提示AS在未来HD疾病的新型分子治疗研发中可能发挥重要作用。
综上,AS在大脑发育和神经精神疾病中均发挥关键作用。AS的发生受多种剪接因子的调控,并且不同剪接因子具有组织特异性,所以在各种组织区域调控不同的AS事件。当这些剪接因子发生异常时,可直接影响AS的模式,从而影响生物过程。在神经系统中,剪接因子调控的AS模式发生变化,直接影响到神经祖细胞增殖、迁移、分化和突触形成等过程,从而产生各种神经系统相关疾病。除了上文提到的一些剪接相关致病基因之外,AS中关键的RBP同特定的前体mRNA和蛋白质结合形成的高度动态化RNP复合物,在调节剪接、编辑、聚腺苷酸化、核输出、定位、翻译和稳定性方面发挥关键调节作用。近年来大量研究表明,多种剪接调节因子在不同神经系统疾病中均扮演着十分重要的角色(表1)。

表1 部分剪接调节因子与神经系统疾病
5 结语
在神经发育过程中,剪接过程异常改变可导致神经发育相关疾病的发生。随着反义寡核苷酸和基因编辑等技术的高速发展,有望改变人类遗传变异和疾病中常见突变的影响。对神经退行性疾病中涉及AS机制的研究,有望为新的治疗方案研发拓展出新视角,并为这种目前仍然无法治愈疾病的诊治带来希望。另一方面,随着近年来单细胞和高通量测序技术的不断突破,有望将AS同更多的神经精神疾病联系起来,为阐明其发病机制和研发干预措施提供新线索。而冷冻电子显微镜和图像采集的新突破,将有望加速提升对剪接体高分辨率结构的解析和理解。随着对AS调控机制的深入理解,将有望创造出新的疾病治疗策略,研发出个性化药物,为临床包括脑发育与神经精神疾病在内多种疾病的诊疗提供全新的解决方案。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29 01:57:52
保健医苑(2022年4期)2022-05-05 06:11:14
上海金属(2021年6期)2021-12-02 10:47:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
中国生殖健康(2020年4期)2021-01-18 02:58:10
生物学通报(2019年3期)2019-02-17 18:03:58
中国生殖健康(2018年4期)2018-11-06 07:12:16
中国卫生标准管理(2015年18期)2016-01-20 09:27:01
湖南中医药大学学报(2015年3期)2016-01-06 01:34:04
食管疾病(2015年3期)2015-12-05 01:45:09
