
儿童原发性1型高草酸尿症一例报道并文献复习
2021-01-14 05:24周旭东赵兴华许长宝李武学赵永立
中国全科医学 2021年9期
周旭东,赵兴华,许长宝,李武学,赵永立
原发性1型高草酸尿症(PH1)是一种常染色体隐性遗传病。因AGXT基因发生突变,导致其编码的肝脏特异性丙氨酸乙醛酸转化酶(AGT)表达缺陷,乙醛酸转氨生成甘氨酸减少,氧化生成草酸增加,草酸盐沉积于肾脏及多个肾外器官,最终引发多发性和复发性草酸钙结石、肾钙质沉着症[1]。由于临床对PH1认识不足,常导致误诊、漏诊,患者就诊时常已进展为终末期肾病(ESRD)。本文报道了1例PH1患儿,其于郑州大学第二附属医院初次就诊时进行代谢评估,尿草酸水平>100 mg/24 h,排除其他疾病后考虑PH的诊断,并进一步通过AGXT基因测序明确诊断为PH1,目前正在进行为期6个月的干预治疗及密切随访,现报道如下。
专家点评:
由于临床对原发性1型高草酸尿症(PH1)的认识不足而常导致误诊和漏诊,不能及时或恰当治疗,患儿常早期进展为终末期肾病。本文通过对1例6岁尿结石患儿的诊治过程进行总结分析,提示针对儿童肾结石需要进行全面的代谢评估,以帮助查找遗传病因,做到精准诊断及进一步的临床治疗和遗传咨询服务,对临床相关病例有提示作用。
(首都医科大学附属北京妇产医院 孔元原)
1 病例简介
患儿,男,6岁,汉族,首发症状为排尿时突发尿痛,可见结石排出,2019-02-12至河南省平顶山市宝丰县人民医院行彩超检查示:双肾结石、右侧输尿管结石、右肾积水、膀胱结石。后为进一步治疗于2019-02-22入住郑州大学第二附属医院泌尿外科,期间未行特殊治疗。患儿既往可见排尿突然中断症状,生长发育正常。家族史:患儿父母非近亲结婚,父系及母系三代无相同病史。
入院查体及辅助检查:发育正常,营养中等,智力正常,身高130 cm,体质量24 kg;血压105/77 mm Hg(1 mm Hg=0.133 kPa);右肾区压痛、叩击痛,未发现其他阳性体征。CT检查结果:双肾结石,右侧输尿管末端结石并右肾积水,膀胱结石(见图1)。
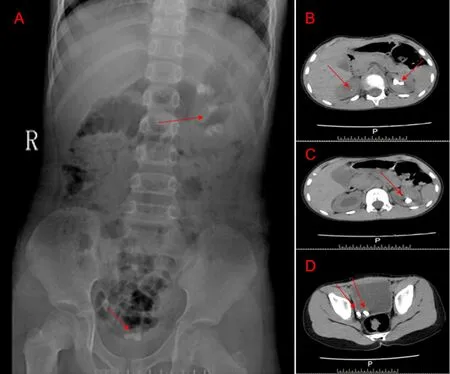
图1 患儿影像学检查结果Figure 1 Imaging findings of the male child with primary hyperoxaluria type 1
实验室检查:血常规示白细胞计数5.9×109/L,血红蛋白133 g/L,血小板403×109/L;尿常规示pH值6.0,红细胞沉降率(+),尿蛋白阴性;血生化检查示血尿酸385 μmol/L,血尿素氮4.56 mmol/L,血肌酐40 μmol/L,甲状旁腺素35.6 ng/L。患儿共行6次24 h尿液成分分析,结果见图2。结石成分分析为一水草酸钙>95%。
基因测序分析:对患儿及其父母进行基因测序,患儿基因测序在AGXT基因第9外显子上发现突变(c.864G>A:p.Trp288X),第2外显子上亦发现突变(c.346G>A:p.Gly116Arg)。患儿母亲基因测序检测到AGXT基因第9外显子突变(c.864G>A:p.Trp288X),患儿父亲基因测序检测到AGXT基因第2外显子突变(c.346G>A:p.Gly116Arg)。根据美国医学遗传学与基因组学会(ACMG)的序列变异解释标准和准则[2],突变(c.864G>A:p.Trp288X)为无义突变,可能为AGXT的致病变异;突变(c.346G>A:p.Gly116Arg)是来自患儿父亲的错义突变,其存在于转氨酶的一个结构域,对AGT蛋白的功能很重要(见图3)。本研究发现的两个AGXT基因突变均为杂合突变,在氨基酸水平上,无义突变导致停止扩增而不是色氨酸形成(W288X),而错义突变导致精氨酸形成而不是甘氨酸(G116R)。
诊断及治疗:患儿的临床表现符合PH1的临床特点,结石成分分析为一水草酸钙>95%,AGXT基因测序为复合杂合突变,临床最终诊断为PH1。患儿在本院的治疗分为3个阶段:第1阶段,嘱患儿出院后大量饮水〔3 L·(m2)-1·d-1〕,口服维生素B6(5 mg·kg-1·d-1)以抑制草酸形成,并口服枸橼酸钾(0.1 mg·kg-1·d-1,早、中、晚3次服用)以碱化尿液并减少尿钙排泄,抑制尿结石的形成;第2阶段,在第1阶段治疗基础上加用草酸降解酶30 g/d(早、中、晚3次温水冲服),以降解草酸;第三阶段,大量饮水〔3 L·(m2)-1·d-1〕,增加口服维生素B6剂量(10 mg·kg-1·d-1)以抑制草酸形成,并继续口服枸橼酸钾(0.1 mg·kg-1·d-1,早、中、晚服用)以碱化尿液并减少尿钙排泄,抑制尿结石的形成。
随访:由于患儿属于早期发现的PH1,并积极配合治疗,因此本院拟定的随访计划如下:复查血常规、尿常规、肝功能、肾功能、电解质,4周/次,以防止发生泌尿系感染,同时监测肝、肾功能;复查低剂量泌尿系CT(平扫),4周/次,观察患儿有无新发结石、肾钙质沉积等;复查24 h尿液成分,4周/次,观察患儿草酸、枸橼酸、电解质水平,并根据患儿24 h尿液成分分析结果调整用药。随访期间患儿血常规、尿常规、肝功能、肾功能、电解质水平均在参考范围内。
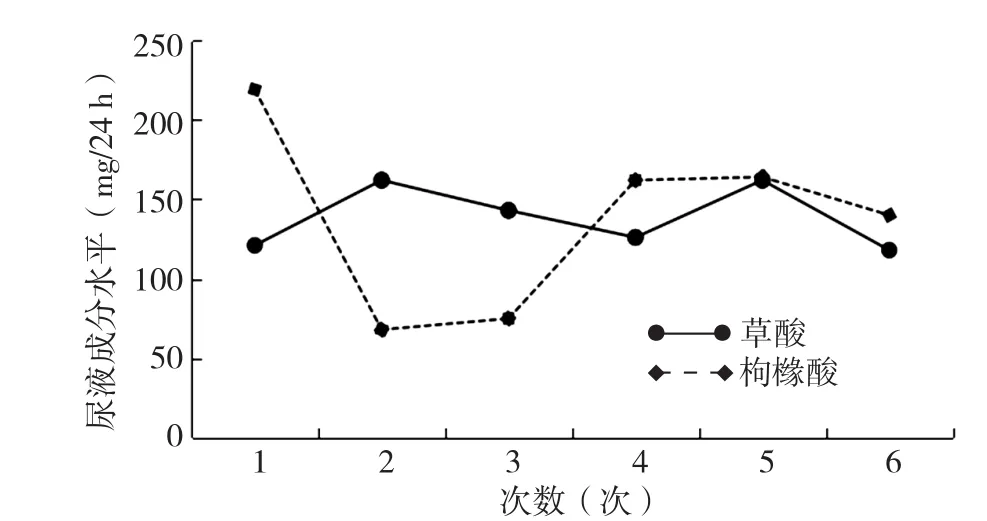
图2 患儿6次24 h尿液成分分析结果Figure 2 Results of 6 times of 24-hour urine composition analysis of this male child with primary hyperoxaluria type 1

图3 患儿及其父母Sanger测序显示AGXT基因突变Figure 3 Sanger sequencing of this male child patient and his parents revealed AGXT gene mutation
2 讨论
原发性高草酸尿症(PH)是一种常染色体隐性遗传病。根据不同的基因突变位点可将PH分为3种亚型,其中PH1是由AGXT基因突变引起的,可导致维生素B6依赖的AGT表达缺陷[3];PH2是由GRHPR基因突变所导致的乙醛酸/羟基丙酮酸还原酶功能障碍[4];PH3是由HOGA1基因突变引起的,该基因编码线粒体4-羟基-2-酮戊二酸醛缩酶[5]。PH1是发病率最高的一种亚型,约占PH的80%[6]。
在中欧地区,活产婴儿中PH1的发病率为1/12万[7];在北非和加里亚群岛,PH1的发病率更高,这可能是由于近亲结婚造成的[8];在欧洲和美国,PH1占儿童和青少年ESRD的比例不到1%[9]。PH1真正的发病原因不清楚,并且是否存在地域性差异仍不确定,但早期的观察研究还是很有意义的[10]。
人体内草酸不能被代谢吸收,只能作为代谢终产物从肾脏排出。草酸盐可以在肾小球内自由滤过,也可以由肾小管分泌。在PH1患者中,由于内源性草酸产生过多,草酸盐在尿液中过饱和,导致肾小管管腔内形成晶体和复合物,而一些晶体会聚集于肾小管细胞表面,最终形成肾结石;另一些晶体会被吸收进肾小管细胞中,并逐渐迁移到肾间质,最终形成肾钙质沉着症。不断进展的肾钙质沉着症和反复发作的泌尿系结石会引起泌尿系感染和梗阻性肾病等并发症。当肾功能进一步损害、估算肾小球滤过率(eGFR)下降至30~40 ml·min-1·(1.73 m2)-1时,草酸盐不能通过肾脏完全排出体外,血浆草酸盐浓度逐渐升高,并迅速达到30 μmol/L。草酸会沉积于各种肾外组织器官,包括心肌、骨、皮肤、血管壁、视网膜、中枢神经系统等,长期影响包括心肌病、骨痛、骨钙化、抗药性贫血、皮肤溃疡、血管病变、视网膜病变、周围神经病变等在内的疾病[11]。
临床早期诊断PH1至关重要,便于早期开始干预治疗。对于泌尿系结石患者,任何年龄段均应行24 h尿液成分分析。PH1患者的代谢指标是在无高草酸尿其他病因情况下,尿草酸水平>50 mg/24 h。目前原发性与继发性高草酸尿症没有明确的界限,尿草酸水平>70 mg/24 h通常认为是代谢因素所致,但也有一些继发性疾病,如克罗恩病、短肠综合征、胰腺功能不全等疾病患者尿草酸水平可以>100 mg/24 h[12]。尿草酸水平在50~70 mg/24 h也不能排除PH,如果患者年龄较小、有复发性肾结石、肾钙质沉着症则应行进一步检查。如果肾功能正常,血浆草酸水平对诊断没有帮助,但随着eGFR的下降,血浆草酸水平会上升。血浆草酸水平>100 μmol/L可能来源于PH1,但目前没有明确的方法来区分是PH1造成还是肾衰竭造成。结石成分中一水草酸钙>95%是PH1患者的另一重要特征[13]。肝穿刺活检是诊断PH1的传统方法,也是诊断PH1的金标准[14],但肝穿刺活检为有创性检查,大多数患者不能接受。分子基因检测已经逐渐取代肝穿刺活检成为首选的PH1诊断方法,目前已经发现PH1患者的AGXT基因至少有178个突变位点[15],其中错义突变最为常见;AGXT外显子及其剪切区直接测序是诊断PH1的重要和有效手段[13],存在致病纯合或者杂合突变可以确诊[16]。随着分子生物学和基因检测技术的发展,PH1虽可以明确诊断,但是PH1的早期筛查仍至关重要。
本例患儿6岁发病,以发现泌尿系结石就诊于本院,尿液成分分析提示尿草酸水平>100 mg/24 h,结石成分分析为一水草酸钙>95%,考虑患儿处于PH1高发病年龄段,且各项检查结果均比较典型,因此高度怀疑患儿为PH1,遂进一步行基因测序,结果提示患儿存在引起PH1的两个AGXT基因突变(c.346G>A:p.Gly116Arg;c.864G>A:p.Trp288X),其中一个突变为新的无义突变(c.864G>A:p.Trp288X),而另一个突变(c.864G>A:p.Trp288X)为错义突变。本次对患儿进行基因测序检测到的这种新的无义突变可能对中国PH1患儿的治疗有重要意义。结合患儿的临床表现和基因测序结果,PH1诊断明确,无须再行肝穿刺检查。
关于PH1的治疗,目前主要是根据PH1患者疾病发展阶段执行个体化治疗。在PH1发现早期以保守治疗为主,包括大量饮水〔2~3 L·(m2)-1·d-1〕以防止草酸过饱和[17];口服维生素B6以减少草酸生物合成,推荐剂量为初始5 mg·kg-1·d-1,逐渐增加剂量但不超过20 mg·kg-1·d-1,且至少使用3 个月[17];口服枸橼酸盐(100~150 mg·kg-1·d-1,早、中、晚服用)以碱化尿液并减少尿钙排泄[18]。当患者出现透析指征但又不能行肝肾联合移植时,推荐高效透析,血液透析、腹膜透析或者两者联合以及高通量透析均可[19]。随着PH1病情进展,肾功能进一步恶化,到慢性肾脏病4、5期时,现有的透析方案已经不能快速清除患者体内产生的过量草酸,因此,器官移植成为治疗该疾病的另一种选择,目前肝肾联合移植的疗效最好[19],不推荐单独的肾移植,因为单独的肾移植具有较高的复发风险[20]。近年来,随着分子生物学技术以及基因技术的发展,关于PH1的治疗方案也层出不穷。利用肝细胞移植可以恢复AGT活性[21];由于PH1是单基因致病,重组基因疗法寻找替代酶也可用于该病的治疗[22]。未来可以通过联合各种治疗方案、早期干预以减少PH1患者肝脏草酸的产生、草酸在肾脏以及肾外器官的沉积,减轻患者痛苦,避免行器官移植术[23]。本例患儿属于PH1早期,给予排石治疗后开始进行针对PH1的个体化治疗方案,包括大量饮水、口服枸橼酸钾以碱化尿液并减少尿钙排泄、逐渐增加维生素B6剂量以减少草酸生物合成,并且在治疗过程中尝试使用草酸降解酶以降解草酸,后发现草酸降解酶对该患儿降解草酸无明显作用,遂暂停应用草酸降解酶。在患儿治疗及随访过程中,其肾功能指标均在参考范围内,由于患儿的依从性较差,枸橼酸盐未按照要求服用,复查24 h尿液成分中枸橼酸水平波动较大,但随访期间24 h尿液成分中草酸水平波动在30%以内。
综上所述,对于儿童时期发现的泌尿系结石,均应行全面的代谢评估,当24 h尿草酸水平高于参考范围上限(即>50 mg/24 h),排除肠源性疾病后均应考虑到PH[19]。AGXT基因测序目前已经取代肝穿刺活检成为诊断PH1的首选方法,应注意对已经确诊的患者家属进行基因测序[20]。一旦基因测序诊断确诊为PH1,应该积极制定个性化的治疗方案,并密切随访;如果患者的肾功能已经下降,则应该考虑合适的器官移植方案[6]。
作者贡献:周旭东进行文章的构思与设计、可行性分析,并负责论文撰写;周旭东、李武学、赵永立负责资料的收集与整理;赵兴华负责论文质量控制及校审,对文章整体负责、监督管理;许长宝负责论文的修订和质量控制。
本文无利益冲突。
猜你喜欢
基层中医药(2022年7期)2022-11-17
军事文摘(2022年16期)2022-08-24
基础医学与临床(2022年1期)2022-01-21
时代英语·高一(2018年4期)2018-09-14
医药前沿(2018年2期)2018-01-17
材料科学与工程学报(2016年4期)2017-01-15
饮食科学(2016年3期)2016-07-04
饮食科学(2016年3期)2016-07-04
现代检验医学杂志(2015年2期)2015-02-06
食品工业科技(2014年13期)2014-03-11
