
成年17α羟化酶缺陷症二例临床特征及诊疗分析
2021-01-14 05:24刘君静任丽张卫游志清程莹郎红梅郭蔚
中国全科医学 2021年9期
刘君静,任丽,张卫,游志清,程莹,郎红梅,郭蔚
先天性肾上腺皮质增生(congenital adrenal hyperplasia,CAH)是一种常染色体隐性遗传病,主要由参与肾上腺类固醇激素合成所需酶的先天缺陷引起,不同基因型可诱导不同的表型,其中21-羟化酶缺乏最常见,占所有类型CAH的90%~95%。17α羟化酶缺陷症(17α-hydroxylase deficiency,17OHD)是一种罕见的CAH类型,由位于10q24.3染色体上的CYP17A1基因突变引起[1],估计发病率为1/50 000~100 000,约占所有CAH的1%[1-2]。本研究分析了2例成年17OHD患者的临床特征及诊疗经过,旨在增强临床医生对CAH及其亚型17OHD的认识,现报道如下。
1 病例简介
1.1 病例1 社会性别:女,24岁,主因“反复头昏、乏力11年”而于2013-12-27入住西部战区总医院内分泌科。患者13岁时因乏力、瘫软而发现血钾降低,经补钾治疗后好转,此后每年需静脉补钾,平素稍觉四肢无力时口服氯化钾注射液即可消失;18岁时因头痛而发现血压升高,最高达210/100 mm Hg(1 mm Hg=0.133 kPa),平素间断口服降压药物(具体不详),血压控制在140~150/70~100 mm Hg。患者为顺产,出生时体质量正常,生长过程中身高、体质量、智力与同龄人无明显差异;无月经来潮,未婚未育。患者父母体健、非近亲结婚,育有两女;患者妹妹身体健康。查体:体温为37.2 ℃,脉搏为70 次/min,呼吸频率为16次/min,血压为171/116 mm Hg,身高为161 cm,体质量为52 kg,体质指数(BMI)为20.1 kg/m2;体型偏瘦,全身皮肤偏黑,无明显色素沉着,皮下脂肪少,乳房无发育、无乳晕及色素沉着,心、肺、腹无明显异常,无阴毛、腋毛分布,外阴幼稚,阴蒂肥大,双下肢无水肿。
1.2 病例2 社会性别:女,31岁,主因“血压升高5年余,肌肉酸痛、乏力2周”而于2019-02-28入住西部战区总医院内分泌科。患者于5年前在当地医院就诊并发现血压升高,最高达186/110 mm Hg,平素未服用降压药物,自测收缩压波动于140~150 mm Hg,因无明显不适而未进一步诊治。患者入院1周前因乏力、肌肉酸痛至当地医院就诊,查血压偏高(具体不详),行电解质检查发现血钾为1.6 mmol/L,予以补钾、降压等治疗后症状好转,复查血钾逐步恢复正常,住院期间行腹部CT发现双侧肾上腺占位性改变并建议至上级医院进一步诊治,遂就诊于西部战区总医院内分泌科。既往史:患者10余年前因月经未来潮而于当地医院检查并诊断为“先天性始基子宫”,4年前因手部创伤而发现“骨骺未闭合”,均未进一步诊治。患者为弃儿,养母诉其幼时瘦小,但生长过程中身高、体质量、智力与同龄人无明显差异;24岁结婚,配偶体健,未生育。家族史不详。查体:体温为36.3 ℃,脉搏为104次/min,呼吸频率为20次/min,血压为131/93 mm Hg,身高为168 cm,体质量为51.4 kg,BMI为18.2 kg/m2;体形消瘦,四肢细长,全身皮肤无色素沉着,气管居中,未见喉结,甲状腺不大,乳房未发育、无乳晕,心、肺、腹无明显异常,无阴毛、腋毛分布,阴蒂肥大,双侧腹股沟区可触及一包块、无压痛、活动度可,双下肢无水肿。
1.3 实验室及影像学检查 2例患者均在住院期间完善实验室检查,包括血常规、尿常规、便常规、生化指标(肝肾功能指标、血糖、血脂指标、电解质)、24 h尿电解质、性激素、血浆皮质醇及促肾上腺皮质激素(ACTH)节律、24 h尿游离皮质醇、垂体激素、醛固酮、肾素活性、甲状腺功能、儿茶酚胺等,并行肾上腺CT、妇科超声检查,病例2还行双手X线及骨密度检查。
1.4 基因测序、染色体核型检测 病例1乙二胺四乙酸(EDTA)抗凝外周血外送至金域检验进行17OHD和/或17,20裂解酶缺陷症CYP17A1基因测序及外周血染色体核型检测;病例2 EDTA抗凝外周血外送至智因东方进行先证者全外显子测序,并自行前往四川大学华西医院进行外周血染色体核型检测。
本文要点:
17α羟化酶缺陷症在临床上较为罕见,主要表现为不同程度的低钾血症、高血压、性腺发育及性征异常、肾上腺增生。本研究中2例患者分别以低钾血症、高血压起病,而由于低钾血症、高血压在临床上并不少见且经对症处理可较快缓解,因此初诊时极易忽视对其病因的探索,之后依据病情迁延、反复及性发育异常虽可逐步确诊,但确诊时间常会延迟至初次发病后数年,甚至成年后才能被确诊。17α羟化酶缺陷症患者长期存在的病理生理改变及性发育异常除会对器官功能造成影响外,还会带来很多心理问题及社会问题,因此需早期诊断、及时干预以达到改善预后、提高生活质量的目的。
2 结果
2.1 临床表现 2例患者社会性别均为女性,青春期无月经来潮,伴性腺发育异常且无第二性征发育,并均以低钾血症、高血压为主要表现而住院诊治;2例患者外观均为四肢细长、体型偏瘦,缺乏女性柔和气质,其中病例2双侧腹股沟区发现疑似隐睾包块。2例患者成年后明确诊断年龄分别为24岁、31岁。
2.2 实验室检查结果 2例患者24 h尿钾、卵泡期促卵泡生成素、卵泡期黄体激素、促肾上腺皮质激素均升高,血钾、雌二醇、孕酮、硫酸脱氢表雄酮、血浆皮质醇、17羟孕酮(仅病例2)、24 h尿游离皮质醇均不同程度降低,睾酮均在参考范围内,详见表1;病例1醛固酮正常,病例2高醛固酮低肾素活性、醛固酮/肾素比值明显升高,进一步行0.9%氯化钠溶液负荷试验发现负荷后醛固酮为256.8 ng/L、醛固酮未被抑制在50.0 ng/L内。2例患者血、尿、便常规及肝肾功能指标、血糖、血脂指标、甲状腺功能指标、儿茶酚胺、垂体激素均未见明显异常。
2.3 影像学检查结果 2例患者肾上腺CT检查均提示肾上腺存在不同程度增生及多发结节样占位性改变(病例1有诊断报告但肾上腺CT检查结果无法获取,病例2肾上腺CT检查结果见图1)。病例1妇科超声检查提示先天性始基子宫、卵巢显示不清,病例2妇科超声检查提示双侧腹股沟区低回声结节、疑似隐睾、未见正常子宫及双附件。病例2双手X线检查提示骨骺未闭合,骨密度检查提示骨量减低。
2.4 基因测序、染色体核型检测结果 2例患者CYP17A1基因均存在2处杂合突变,其中共同变异位点为CYP17A1:c.1459_c.1467delGACTCTTTC,病例1另一变异位点为c.1084C>T,病例2另一变异位点为c.286(exon1)C>T(见图2~3)。病例1染色体核型为46XX,与社会性别一致;病例2染色体核型为46XY,与社会性别不一致。
2.5 治疗及随访 病例1住院期间给予氯化钾缓释片(1 g/次、3次/d)、硝苯地平控释片(30 mg/次、1次/d)、泼尼松(8:00服5.0 mg,16:00服2.5 mg)治疗,血压控制可、血钾恢复正常后出院;出院后口服泼尼松(8:00服5.0 mg,16:00服2.5 mg),2周后复诊无明显不适,血压正常,血钾为4.71 mmol/L,ACTH为 33.6 ng/L,醛固酮为86.80 ng/L,之后失访。病例2住院期间给予哌唑嗪片(1 mg/次、3次/d)、氯化钾缓释片(1 g/次、3次/d),诊断明确且病情稳定后出院;出院后口服醋酸地塞米松(0.25 mg/次、1次/d),1个月后复诊无不适,血压正常,复查血钾为4.06 mmol/L,血钠为 140.00 mmol/L,ACTH为44.9 ng/L,血浆皮质醇为0.43 μg/dl,3个月后进行电话随访,患者诉无不适,复查血钾、血压均正常,遂嘱其尽快行腹股沟包块切除术。

表1 2例成年17OHD患者部分实验室检查结果Table 1 Partial laboratory examination results of the two adults with 17α-hydroxyase deficiency
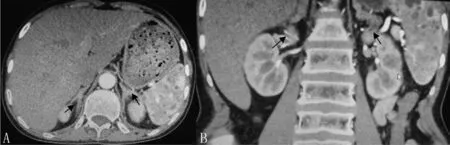
图1 病例2肾上腺CT检查结果Figure 1 CT examination results of adrenal glands of Case 2
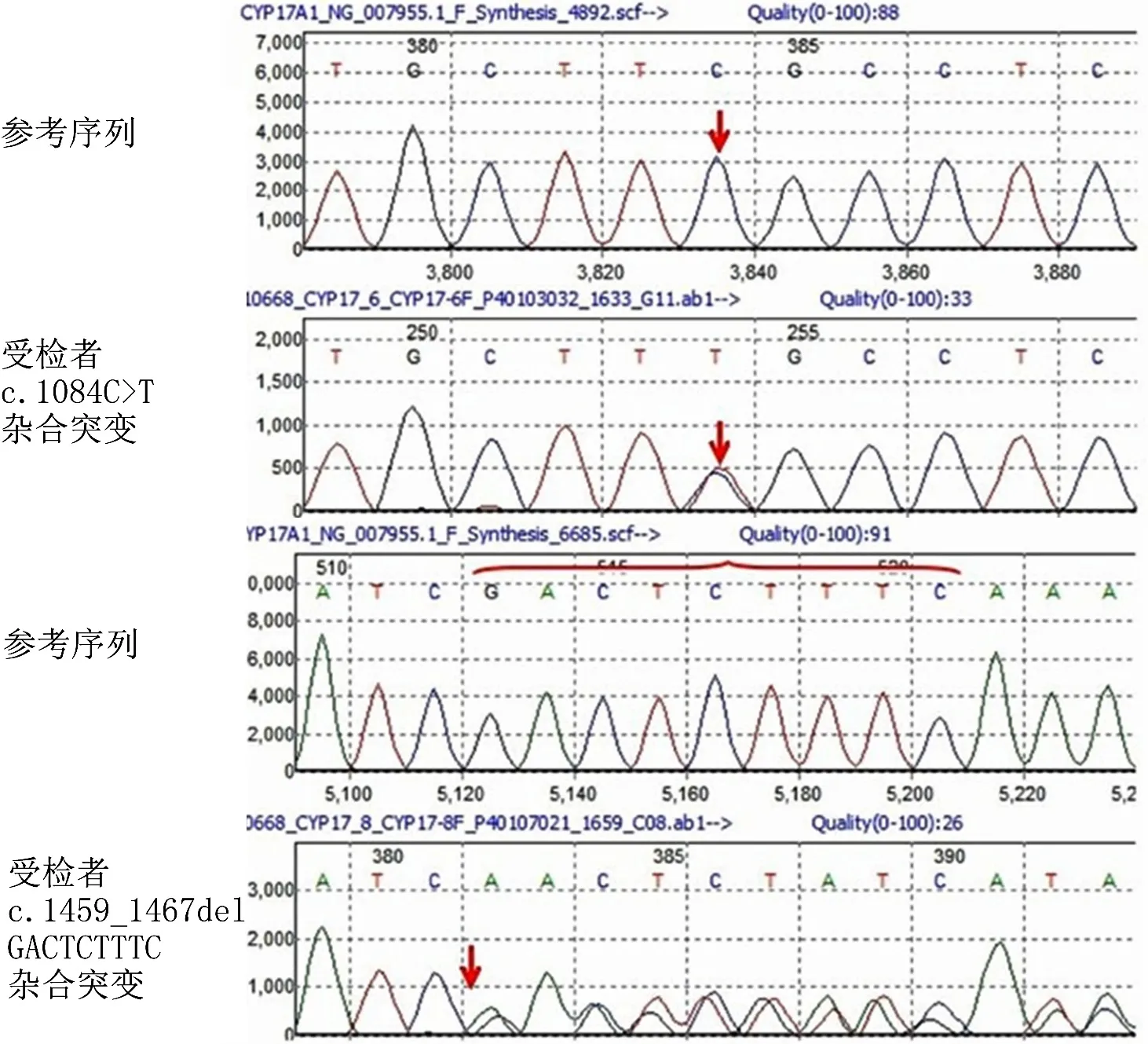
图2 病例1基因测序结果Figure 2 Gene sequencing results of Case 1

图3 病例2基因测序结果Figure 3 Gene sequencing results of Case 2
3 讨论
由于CYP17A1基因主要编码具有活性的17α羟化酶和17,20碳链裂解酶的细胞色素P450c17酶,因此CYP17A1基因突变可表现为17α羟化酶和/或17,20碳链裂解酶缺陷[3],多数患者两种酶常同时存在缺陷[4]。研究表明,类固醇激素合成通路受阻会导致17羟孕酮、11-去氧皮质醇、皮质醇、硫酸脱氢表雄酮、雄烯二酮和睾酮下降[5]及盐皮质激素合成通路增强,进而造成机体产生过剩的11-去氧皮质酮、皮质酮,其中以11-去氧皮质酮升高最为显著。11-去氧皮质酮具有强盐皮质激素作用[6],可导致钠水潴留、钾离子流失并通过抑制肾素-血管紧张素系统而引发低肾素活性,临床表现为高血压、低钾血症;此外,11-去氧皮质酮还具有糖皮质激素和盐皮质激素作用,因此17OHD患者较少出现肾上腺皮质功能低下的临床表现,且几乎不会出现威胁生命的肾上腺危象,但可能导致初次确诊时间延迟。既往报道的17OHD病例醛固酮表现不一致,降低、正常或升高均有报道[7-8],但极少提及原发性醛固酮增多症的确诊试验。本研究中病例1醛固酮正常,病例2醛固酮及醛固酮/肾素比值均明显升高,进一步行0.9%氯化钠溶液负荷试验发现其负荷后醛固酮未被抑制在50.0 ng/L内,达到原发性醛固酮增多症诊断标准,结合其性发育异常及糖皮质激素治疗后临床转归提示17OHD患者可能会出现原发性醛固酮增生症0.9%氯化钠溶液负荷试验阳性。
在性发育方面,由于17OHD可导致肾上腺、性腺中硫酸脱氢表雄酮及雄烯二酮合成障碍并最终引发雌、雄激素水平降低及性征发育异常,因此疑诊该病时应行染色体核型检测。CHORMANSKI等[9]研究指出,染色体核型为46XX的17OHD患者如在青春期前不发生高血压或低钾血症则无不适症状或体检异常,且常因青春期延迟、原发性闭经和缺乏第二性征就诊;染色体核型为46XY的17OHD患者外生殖器表型可从女性到含糊不清或小的男性生殖器,呈男性假两性畸形,如表型为女性则可能有腹股沟疝或腹股沟肿块病史,且在青春期前若未被发现则会与染色体核型为46XX的17OHD患者一样因青春期延迟、原发性闭经和缺乏第二性征而就诊,同时此类患者查体可发现会阴部为盲端,常被误认为阴道但缺乏女性内部生殖器,影像学检查常提示睾丸隐匿或位于腹股沟内。本研究中2例患者社会性别均为女性,第二性征与上述描述一致,染色体核型分别为46XX、46XY,其中病例2外生殖器表型与染色体核型不同,呈男性假两性畸形。
在诊断方面,一项来自巴西的研究结果显示,17OHD确诊前误诊率高,最常误诊为原发性高血压病(55%)、单纯性腺发育不良(35%)及雄激素抵抗综合征(21%);初诊和确诊的中位年龄分别为13.2、16.5岁,平均诊断时间间隔为3.2年[10]。本研究中2例患者均因低钾血症伴高血压就诊,极易误诊为原发性醛固酮增多症。对于青春发育后期第二性征及月经初潮均未出现者,进行肾上腺皮质激素、性激素及肾上腺影像学检查对初步诊断17OHD具有重要作用,但确诊依赖于基因检测,最好对家族成员同时行基因检测,即家系筛查。本研究中2例患者因其家属拒绝或无法联系到亲生父母而未完成家系筛查。需要指出的是,17OHD可由多个或单个基因突变引起,近期有学者总结了在PubMed发表的181例17OHD患者基因突变情况,发现其中70例(38.6%)为c.985_987delinsAA(p.Y329K) 突 变,55例(30.4%) 为c.1459-1467del(p.487_489del) 突 变, 其 余 43例(31%)为其他突变[11]。本研究中2例患者CYP17A1基因均存在2处杂合突变,其中共同变异位点为CYP17A1:c.1459_c.1467delGACTCTTTC,为CYP17A1基因常见突变位点。
在治疗方面,由于17OHD患者体内高水平的皮质酮及去氧皮质酮具有一定糖皮质激素作用,因此患者几乎不会出现肾上腺危象,但患者仍有乏力、精神差、抵抗力差等糖皮质激素缺乏表现,需补充充足的糖皮质激素以改善上述症状,同时需通过抑制ACTH而减少盐皮质激素分泌以纠正低钾血症及高血压,目前常用药物为地塞米松。本研究中2例患者分别服用泼尼松、地塞米松进行治疗,后期随访发现其血压、血钾均恢复正常,高水平的ACTH也下降至参考范围。蒋建发等[12]通过对30例染色体核型为46XY的17OHD患者进行腹腔镜下性腺切除术发现,大部分患者性腺组织位于腹股沟内口处,少部分位于腹股沟管内,术后病理学检查提示为发育不良的睾丸组织,少数患者发现性腺及相关肿瘤。因此,对于明确诊断的染色体核型为46XY的17OHD患者,应尽早行性腺切除术,而对于社会性别选择为女性的染色体核型为46XY及46XX的17OHD患者,可进行雌激素补充治疗。
有研究表明,17OHD患者经周期性雌/孕激素治疗后阴毛和腋毛生长情况与正常成人相似[13],但TURAN等[14]研究发现17OHD患者即使经高剂量雌激素治疗也无法实现完全的乳房发育,推测其乳腺发育受损可能是由高孕酮和低雌激素生物合成导致乳腺组织中雌激素/孕酮失衡所致。有学者对1例17OHD患者采用激素替代治疗诱导子宫内膜成熟、促性腺激素诱导排卵,结果显示应用促性腺激素后虽有卵泡生长和排卵,但子宫内膜厚度不足,推测长期暴露于高水平孕酮可能导致子宫内膜不成熟和子宫内膜对雌激素治疗不敏感[15];但也有研究表明雌激素治疗可有效降低染色体核型为46XY的女性17OHD患者性激素不足引起的预期成年身高过高的可能性[13],提高骨量并减少持续性骨丢失[16]。因此,虽然对17OHD患者进行雌激素治疗很难使其恢复正常女性性征及生育能力,但在一定程度上改善了其女性性征、家庭生活质量、性激素缺乏并减少了糖皮质激素治疗对骨代谢的不良影响。
综上所述,17OHD发病率低,成年后临床表现具有一定异质性,而早期诊断、早期治疗可改善患者成年后生活质量,因此临床医生应加强对该病的认识并提高早期诊断、鉴别诊断水平,以避免误诊、漏诊。
作者贡献:刘君静、游志清进行文章的构思与设计;刘君静、任丽、张卫进行资料及文献整理;刘君静进行论文撰写及修订;游志清、程莹、郭蔚负责文章质量控制及审校,监督管理;郎红梅进行部分论文及英文的修订。
本文无利益冲突。
猜你喜欢
宁夏医学杂志(2020年3期)2021-01-21
透析与人工器官(2020年1期)2020-11-16
中国循环杂志(2020年4期)2020-04-27
养生大世界(2019年12期)2019-12-11
心肺血管病杂志(2019年12期)2019-05-20
家庭科学·新健康(2018年8期)2018-10-30
中国继续医学教育(2016年18期)2016-02-15
哈尔滨医药(2015年2期)2015-12-01
中国当代医药(2015年9期)2015-03-01
医疗装备(2015年14期)2015-02-11
