
文本分析技术在蛋白质生物信息学中应用的案例综述
2021-01-12 09:30苏绍玉鄢仁祥
生物信息学 2020年4期
苏绍玉,徐 婧,鄢仁祥*
(1.福建省科学技术信息研究所, 福州 350003;2.福州大学 生物科学与工程学院,福州 350100)
文本一般是指由一定符号或者文字组成的信息。大部分的文本信息是可以直接人工读取其内容的。文本分析主要指对特定的文本数据的含义进行表示、提取其中特征、并对相应信息知识进行表示和分析的过程。文本分析是文本表示、信息挖掘、信息检索和深入探索的一个基本问题。通常来讲,文本分析是文本数据挖掘、信息检索和大数据分析中的核心技术之一。近些年来,随着大数据、人工智能和机器学习等一系列信息技术的发展,文本分析技术在众多领域得到了越来越多的应用。现今,各行各业包括科研和商业机构都处于信息爆炸产生的海量数据时代,在这种背景之下文本分析技术愈显得重要和亟待发展。
资料或者讯息主要分为视频、音频和文本资料三种类型。其中,文本是多数传统知识的载体形式。近年来随着机器学习中的深度学习(Deep learning)[1]算法的进展,音频和视频的识别率显著提高并得到越来越多的应用。这可以在最近几次的图像识别竞赛结果以及现实生活中各种视音频识别应用中得以体现。近年来在对资料进行分析中的另外一个进展就是文本分析技术。各种机器学习算法广泛地应用于文本的解析之中[2-5],特别是人工神经元网络[6]、随机森林[7]以及支持向量机[8]等主流方法。尤其明显的是在近几年,深度学习算法结合文本分析在不少领域中都有着重要的应用[9]。
文本的分析过程可以简单地分为设计规划、文本信息采集、文本信息解析和文本内涵的应用等环节。图1列出了文本分析的基本通用过程。这个基本过程主要就是文本数据的收集、统计分析建模、挖掘其中潜在知识、以及应用挖掘到的知识等过程。文本数据的收集一般可以从互联网(例如,使用爬虫程序或者直接下载)、各种数据库、现存的文件或者实际正在开展的调研项目中获取;之后采用统计模型或者机器学习算法等对数据进行深入分析以及建模;挖掘出来的知识一般以图表或者一些特定的模型形式表示;最终,经验证的可靠知识和规律则可以直接应用于实际之中。
1 文本分析技术在生物信息学中的应用
文本分析的方法已经广泛应用于蛋白质的研究之中。蛋白质相关的计算研究是生物信息学中的一个重要方向。从结构层次的角度来看,蛋白质的一级序列、二级结构、三级结构以及功能等信息都可以用文本进行表示(本文将会对蛋白质的四个结构层次进行详细论述见图1)。在生物学中,蛋白质分子广泛地参与细胞中的各项生理和生活过程,包括生长、发育、生殖、代谢和免疫调节等各个环节,并与各种重要疾病的发生、发展和治疗密切相关。同时,蛋白质是一种重要的药物靶标,其相应机理一直受到科学界的极大关注。蛋白质的结构,特别是三维空间构象,对其生物学功能有重要作用。然而,与蛋白质在生物学中的重要性形成强烈反差的是科学界对于其结构、功能以及相应机理的了解非常贫乏。主要原因是通过实验手段来获得相应信息的过程极其困难,同时需要消耗一定的科研经费。在学术界中,各个专业的科学家通常主要从计算模拟和实验验证的两个角度同时进行来系统性地研究蛋白的作用机制。蛋白质结构与功能的研究通常被认为是学术价值和应用价值兼备的研究课题之一,其研究不仅可增强学术界对蛋白质折叠规律的了解,而且开发出的预测方法还可直接用于指导蛋白质工程、功能基因组学和新药研发等实验研究过程。因此,近年来生物信息学的研究中越来越偏重于往蛋白质相关的方向发展。与此同时,在“互联网+”和大数据快速发展的背景下,文本分析技术越来越多的应用于蛋白质分析的研究中,这对生物学领域以及情报分析行业都有重要意义。文本分析研究中有效融合了数据分析和信息处理以挖掘蛋白质相关的有价值的信息,可在生物学研究中发挥引领和导向作用。

图1 文本分析的基本过程Fig.1 Basic procedure for text analysis
传统的信息情报[10]研究工作一般按照任务和项目进行相应的研究。信息情报技术目前已经介入包括经济、社会、科技预测研究和重大智库决策支持研究之中。同时,也包括生物科学的研究,信息情报技术特别是文本分析技术目前也已经得到了较大的应用。情报分析技术的进展需要全面客观理性发现、分析和研究特定领域的需求。科技情报机构一般不仅仅为政府部门提供科技情报服务,也应为科学技术的进步,例如生物学的方法,提供一定的技术储备和应用理论基础。通常来讲,情报信息技术的发展一般涵盖不同时空领域的政治、经济、社会、生物学、人文文化等各方面的信息。特别是近些年来,生物学和信息技术结合得越来越紧密,情报分析中的文本分析技术在这个特定领域中也得到了越来越广泛的应用。我们将在下面段落中对文本分析在生物信息学中蛋白质的计算分析具体应用进行举例。
2 文本分析技术应用于蛋白质折叠类型的识别
蛋白质折叠识别,即从蛋白质序列识别出其三维结构类型,是生物信息学中的一个重要研究方向。蛋白质一级序列结构中一般使用20个字母表示20种标准氨基酸,相应的序列信息以文本文件的形式进行储存。目前学术界可以获得的最大的蛋白质序列数据库为美国国立生物技术信息中心(National Center for Biotechnology Information)[11]的NR(Non-Redundant Protein Sequence Database)数据库。NR数据库不同于一般数据库(例如MySQL和微软SQL server数据库),NR数据库就是采用最简单的文本文件的格式(也称为fasta格式)来储存蛋白质序列。所以不少文本分析的算法可以直接应用于蛋白质的序列分析。
蛋白质折叠识别涉及蛋白质一级序列到三维空间结构类型(即蛋白质三级结构)的映射(见图2),主要使用方法为穿线法(Threading)或者也称为逆折叠法(Inverse folding)[12],意为把目标序列穿过蛋白质的三维空间结构以判断序列与结构之间的匹配程度。匹配程度越高,则相应的打分分数也越高。折叠识别方法的理论基础是蛋白质在序列不太相似的情况下,蛋白质的空间结构仍然可能相似,即蛋白质之间存在弱同源性。这是因为蛋白质在千百万年甚至更长时间的进化历程中,为了保持功能的相对稳定结构也会保持相对稳定,但是序列由于各种突变因素的存在而不断变化。这个特性可以总结为蛋白质的结构比序列保守。所以自然界中存在序列很不相似但是结构相似的蛋白质结构。文本分析方法在蛋白质折叠识别中也有重要应用。其中代表性的方法之一为Cheng和Baldi提出的一种基于文本分析和机器学习的方法。在这种方法中,作者主要采用文本分析中信息检索(Information retrieval)[13]的方法。给定一个查询蛋白质的一级结构序列,折叠的目标识别是将所有可能的模板按照它们序列与结构相关性排序,这个过程类似于谷歌和其他搜索引擎对与用户查询关联的网页进行排名。在这样的分析算法中,越相似的蛋白质理论上也就越容易排名比较靠前。在这个案例中,文本分析技术是一种总体的策略,即采用类似于搜索引擎算法的策略把蛋白质的折叠类型给检索出来。在具体的参数构建中(即机器学习中输入的特征向量),这个研究具体采用可以表征蛋白质序列和结构相似性的各种指标。
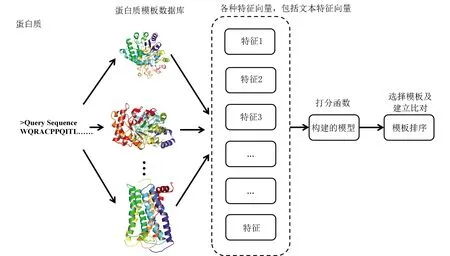
图2 蛋白质折叠识别的基本流程Fig.2 Basic flow for protein fold recognition
文本与蛋白质的三维空间有着紧密关系。虽然蛋白质的结构可以用三维图形的形式显示,但是存储蛋白质结构的文件却是文本文件。这个文件一般是以PDB的格式进行存储的。目前PDB数据库(http://www.rcsb.org/)中存储的生物大分子结构数据量有16万多个,其绝大多数是蛋白质的结构文件。因为蛋白质的结构文件采用文本形式存储,所以文本分析技术也广泛应用于蛋白质结构分析中,包括信息的提取、分析以及结构优化等。
3 蛋白质二级结构的预测
在生物学中,蛋白质的二级结构除了用图形结构表示外,同时也可以采用本文的形式进行储存。在蛋白质二级结构中,学术界一般用字母H代表螺旋、E代表折叠以及C代表无规则卷曲。蛋白质二级结构预测的经典算法是Psipred[14]。Psipred算法由Jones于20世纪90年代开发,现今仍然有广泛的应用。序列一般通过NCBI PSI-BLAST[15]程序搜索NCBI的NR数据库生成序列谱(Profile)信息,序列谱一般可以反应20种氨基酸出现的频率。表1是使用PSI-BLAST程序对蛋白质进行迭代搜索得到的PSSM矩阵,即蛋白质每个位置20种氨基酸出现的频率。研究人员可以进步一地使用人工神经元网络(Neural network)算法来对序列谱信息进行建模,并准确预测出相应蛋白质每个氨基酸的二级结构。有时也会测试主流的深度学习算法,例如Tensorflow[16]等在相应特征相应上的预测性能。在实际研究中,科研人员一般构建一系列有效的特征向量,用于准确地预测未知蛋白的二级结构。这样面对一个新的蛋白质,其二级结构信息将准确地展示在科研人员面前,这为进一步的科研提供了非常有用的拓扑结构信息。目前主流的蛋白质二级结构测预测精度一般可以达到80%以上。图3列出了常见的二级结构程序所使用的算法框架。
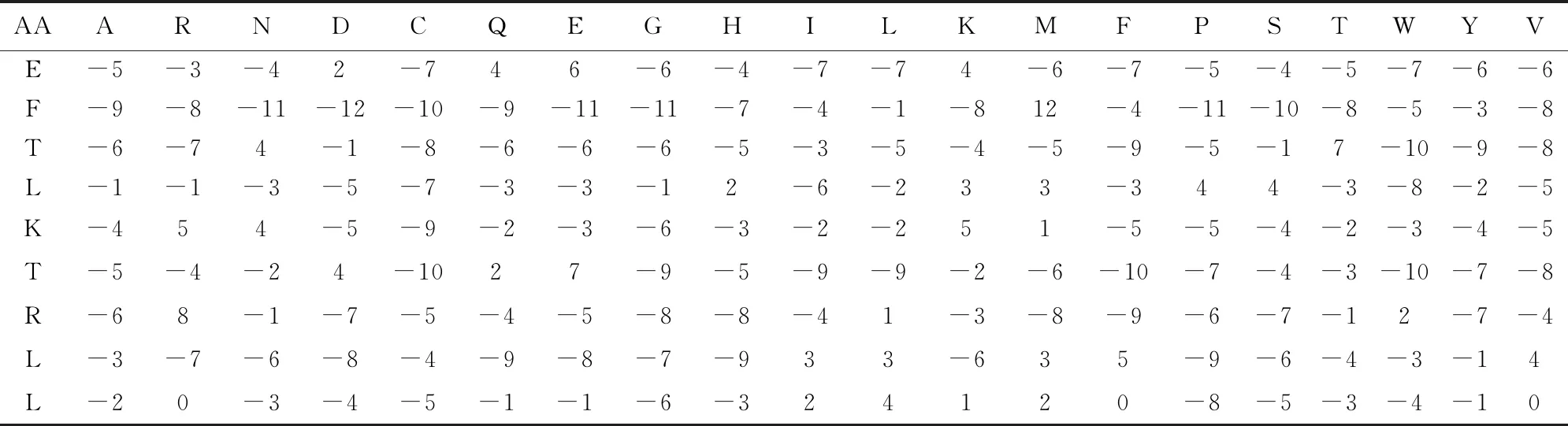
表1 4ay7[17]蛋白质前9个氨基酸的PSSM矩阵Table 1 PSSM matrix of the top 9 amino acids of protein 4ay7[17]
根据形态以及功能特点,生物体内的蛋白可以分成纤维状蛋白(Fibrous protein)、球状蛋白(Globular protein)和膜蛋白(Membrane protein)三大类。有一些特定的蛋白质,例如膜蛋白,其二级结构预测准确率相对较低。这可能是因为膜蛋白处在生物膜之中,膜蛋白的跨膜区与非跨膜区的区别以及功能作用差异是比较大的。这使得膜蛋白有着与球蛋白不同的生物化学特性。准确地获得膜蛋白跨膜区与非跨膜区的信息将有时对判断膜蛋白的生物学功能起到关键的决定性作用[18]。因此,开发针对膜蛋白的二级结构预测算法也是未来的一个重要方向。
4 基于互联网数据库的蛋白质序列功能信息解析
从新测序的蛋白质组和基因组序列中准确识别出目标蛋白是现代组数据功能注释的重要步骤之一。自从各种基因组和蛋白质组测序计划成功开展起,各种测序技术得到极大的发展。因此,大量的物种随后相继被测序。现在几乎每天都会产生大量的基因和蛋白质序列[19]。而新测序的基因和蛋白质序列中一部分数据一般是没有注释功能的。如何从一个新测序物种筛选出潜在的目标基因和蛋白质是科学家们亟待面对的重要的工作之一,这同时也是研究人员判断一个后续研究项目的重要性和是否开展一个研究项目的关键科学依据。现代蛋白质组数据的特点是序列多样性复杂、以及数据量大等特点。首先,科研人员通常研发一系列创新性和使用的算法[20]。新算法可以从全基因组规模的数据中识别出蛋白质相互作用网络,以及预测蛋白质三维结构和药物配体结合模式。同时,一系列创新性的编码及模型算法方式将用于构建针对特定目标蛋白的生物信息学模型[21]。得到计算模型后,研究人员通常将进行相应的蛋白质组学实验对模型进行验证和进一步研究。

图3 基于文本分析和机器学习的蛋白质二级结构预测Fig.3 Prediction of protein secondary structure using text analysis and machine learning
图4列出了常见以蛋白质序列为起始的功能注释流程。在蛋白质功能研究中,科研人员通常开发能适应整个蛋白质组规模的目标蛋白识别的算法。将在保证预测准确定性的前提下,优化程序让程序的运行速度更快。相应算法可以直接应用于蛋白质组注释,以及筛选出重要的目标蛋白,这为基因组科学家们相应的后续研究提供了强有力的计算支持。在这个过程中,各种算法包括文本分析方法得到广泛的应用。
5 文本分析技术在蛋白质生物信息学中的其他应用
在以上提到的几个例子之外,文本分析技术在蛋白质生物信息学的其他方面也有着重要应用。Ross等人提出,在传统构建进化树的方法上,也可以结合文本分析和同源性方法进行构建[22]。Ross等人提出的方法扩充了文本的表示以及应用领域,其方法也有助于生物科研界对文本数据和生物进化之间的理解。论文文献本身就是一种文本形式。Hassani等人使用文本分析技术,系统性搜索现有医学文献数据。基于此种方法,Hassani等人据创建与植物胁迫反应有关的拟南芥蛋白质的知识库。在构建的知识库的基础上,提出多种评分指标来识别关键的蛋白质-胁迫关联[23]。Pavlopoulos等人开发出来一种名为OnTheFly的在线工具,可以实现对Office、PDF以及普通文本文件的信息提取,构建知识图形和网络结构,这个具体可以运用于生物学数据的分析和模型构建(例如蛋白质相互作用网络的构建)。
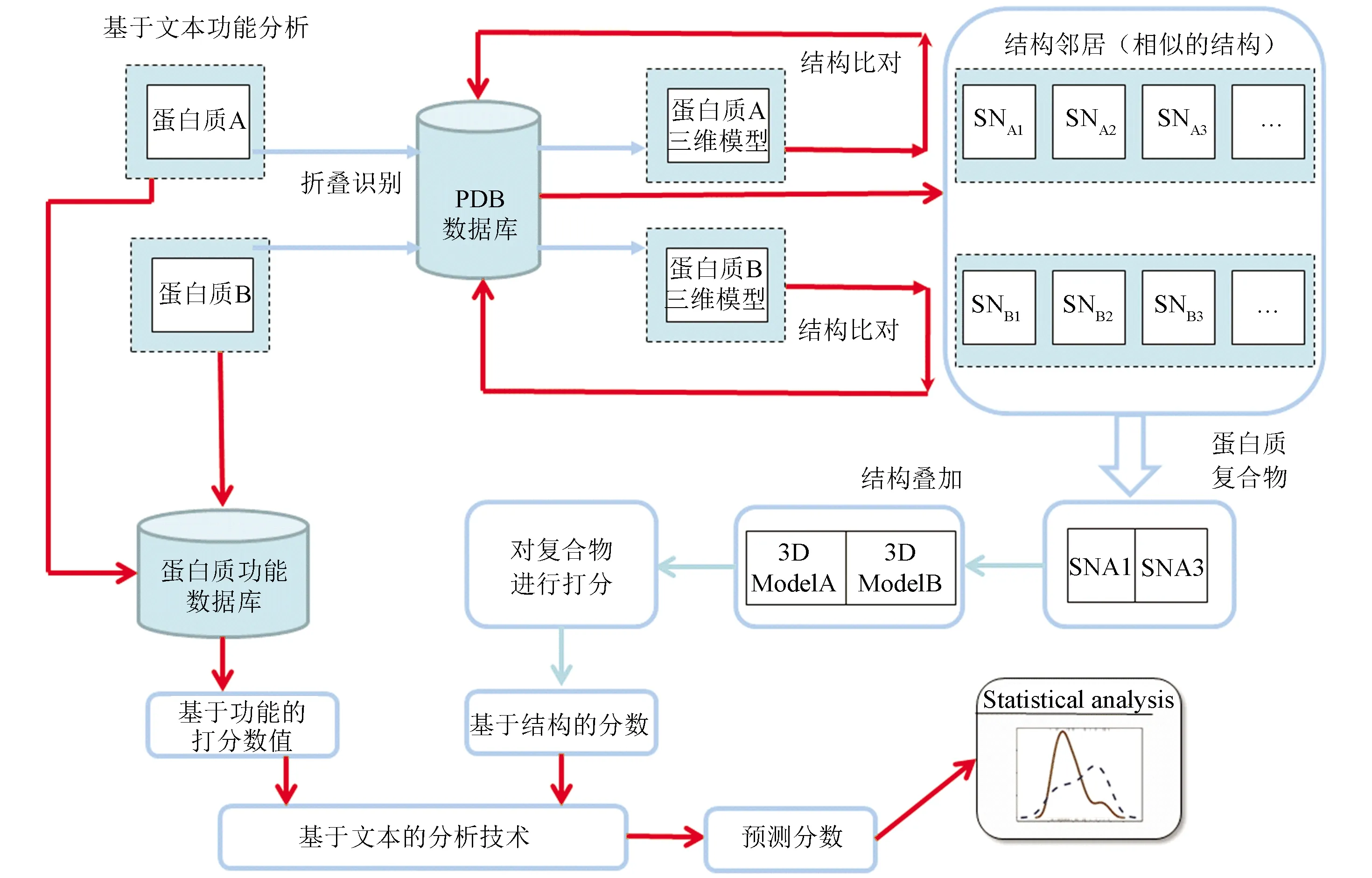
图4 基于蛋白质结构与功能数据库的蛋白质功能预测模型Fig.4 Protein function prediction model based on protein structure and function databases
另外,深度学习在蛋白质生物信息学中也有广泛的应用。例如深度学习算法应用于蛋白质的无规则卷曲模型构建[24]。在蛋白质功能位点预测方面(例如磷酸化位点),深度学习算法也有应用实例[25]。近些年来,随着深度学习算法的发展,之前使用传统文本分析方法建立的模型,研究者也尝试对一部分案例通过深度学习算法进行进一步改进。
6 结 语
文本分析技术是开展科技情报研究工作以及其他相关科学研究的技术支撑之一,在新时期决策难度与风险不断加大的情境下,国内各个相关部门和研究机构有必要审时度势,积极研究这种技术并把之应用于政府资料的收集、分析以及科学研究的各个方面。令人欣慰,文本分析技术已经广泛应用于生物信息学领域中,这也体现在国内不少高校的信息技术相关专业科研教学人员也广泛从事生物信息学研究。本文主要探讨了文本技术在蛋白质计算研究中的几个应用实例。在不同领域中的广泛应用,这也进一步促进了文本分析技术的进展。进展的文本分析技术又可以进一步促进其在科技情报分析中的应用。
本文讨论了文本分析在蛋白质计算分析中的几个应用实例以及核心技术内容。在生物信息学中的应用,进一步促进了文本分析技术的进展。特别是在生物序列的编码,表示序列以及结构相似性的方面,进一步拓宽了文本分析的面,同时也增加了文本分析的内涵。科学研究方面,特别是生物学研究方面,通过理论计算模拟和实验过程有助于对相应分子机制的理解。例如,对蛋白质特有的结构特征进行深入研究,进而阐述与蛋白质相关的生物学功能的分子机制,同时也有助于深入理解膜蛋白序列-结构-功能关系的相应分子机理。相应的研究成果能够应用于基因组和蛋白质组规模的数据应用,也可为新型药物靶标的筛选和药物的开发提供帮助。
文本分析技术在生物信息学中的应用远不止在蛋白质方面,在生物基因组信息学方面也有重要应用。一般而言,能把生物学数据转化为文本数据的方面,都有应用文本分析技术的可能,具体是否可以使用可以阅读相应文献或者进行实际研究测试。另外,本文虽然讨论的是文本分析技术在蛋白质生物信息学领域的应用。但是,一些数据是用文本分析方法是不能直接处理的,例如图片、声音以及视频数据等。图像声音以及视频需要使用其他方法进行数据的基础处理。随着计算机技术的发展,有一些文本采用二进制的格式进行存储,这种格式人工无法直接读取,这可能也是文本技术需要面对的一个问题。文本分析技术仍然存在许多尚待改进的方面。例如,如果是动态地获得网络的上的数据?以及如何降低建模预测出案例的假阳性率?以及得到的数据和模型如何实现实时吸取新的文本数据中的知识?构建模型的文本分析方法是否可以进一步优化等?这些也许是科研人员需要思考以及不断提出新方法改进的方向。
猜你喜欢
肝博士(2022年3期)2022-06-30
云南教育·小学教师(2022年4期)2022-05-17
海外星云(2021年9期)2021-10-14
现代畜牧科技(2021年4期)2021-07-21
艺术评论(2020年3期)2020-02-06
制造技术与机床(2019年10期)2019-10-26
中国博物馆(2018年2期)2018-12-05
电子制作(2018年18期)2018-11-14
现代检验医学杂志(2015年4期)2015-02-06
智能系统学报(2013年5期)2013-08-23
