
肺鳞状细胞癌发生的早期标志物及肿瘤预测模型
2021-01-12 09:30尚文慧王晓曦李晓琴
生物信息学 2020年4期
尚文慧,王晓曦,李晓琴,高 斌
(北京工业大学 生命科学与生物工程学院,北京 100124)
肺癌是常见的恶性肿瘤之一,根据世界卫生组织统计,肺癌的发病率和死亡率均居全球恶性肿瘤的首位。肺癌按组织学分类一般分为小细胞肺癌(Small cell lung cancer, SCLC)和非小细胞肺癌(non-small cell lung cancer, NSCLC),NSCLC约占肺癌的80%,肺鳞状细胞癌(Lung squamous cell carcinoma, LUSC)是NSCLC的主要病理类型之一,导致全世界每年约有40万患者死亡[1]。然而,肺癌的发病机制迄今尚不明确,且起病隐匿。早期的肺癌一般没有明显症状,70%的患者临床发现时已是晚期,失去了临床治疗的最佳时机。肺癌的5年的生存率只有8.9%~15%,而Ⅰ期的肺癌术后5年生存率高达80%,这表明患者存活率与疾病的诊断阶段密切相关。因此,早期诊断对于癌症的治疗和预后都起着关键作用。
近年来,随着分子生物学研究的不断深入,对癌症的发病机制的认识也不断加深,分子基因标志物在癌症临床诊断和治疗中的作用日益受到关注。Liu等[2]人发现TRIM28基因可以作为早期非小细胞肺癌转移和预后的标志物,非小细胞肺癌患者中TRIM28的总阳性率为30.4%(138例中42例),早期患者总阳性率为29.9%(97例中29例)。Tseng等[3]研究SLIT2 在肺癌进展中的基因表达水平,结果表明SLIT2 可以抑制肺癌进展,并认为其可能为肺癌治疗及预后的潜在“治疗靶标”。Yu等[4]也报道了PEBP4作为分子标志物与肺鳞状细胞癌的发生发展、浸润转移以及分化有关,LUSC患者中PEBP4的总阳性率达到了93.4%,然而PEBP4依赖肿瘤阶段,早期(I期、II期)患者PEBP4表达明显低于晚期(III期、IV期)患者(p< 0.05)。由于单一的基因标志物对LUSC的检出率不高,并且早期LUSC标志物较少,因此探索新的可靠的分子标志物对LUSC 的诊断和治疗意义重大。
基因表达水平与癌症的发生密切相关。众所周知,产生癌变的因素有很多,包括基因突变,抑癌基因的功能丧失,原癌基因的激活,以及其他与癌症相关的因素。基因表达数据代表了每个基因的即时表达数据,从这些数据中能够挖掘到有用的信息,发现与癌症相关的基因标志物。癌症基因组图谱计划数据库(The cancer genome atlas, TCGA)和基因表达组数据库(Gene expression omnibus, GEO)为研究人员提供了大量的基因表达数据,如何克服高维小样本的特点进行特征基因的挖掘是生物信息学研究的一个难点,现有研究通常运用统计学方法来挖掘与肿瘤相关的基因。李建更等[5]提出一种多步降维法,该方法运用基因表达差异显著性分析方法(SAM)、偏最小二乘VIP系数法(PLS)和基于巴氏距离的顺序前向搜索方法(BD-SFS),最终提取到20个能将胃癌亚型有效分开的特征基因,利用支持向量机作为分类模型,准确率达到89.43%。Zhang等[6]通过迭代降维递归法筛选出67个特征基因,对LUSC癌症Ⅰ期、Ⅱ期和Ⅲ期样本进行分类,准确率达到86.3%。Feng等[7]因表达值筛选得到一组188个基因组成的基因团,能够较准确地区分乳腺原发癌与癌旁的正常乳腺组织。Lau等[8]使用基因表达数据为患者总体生存率建立三基因(STX1A、HIF1A、CCR7)分类器,该分类器能够对非小细胞肺癌Ⅰ期和Ⅱ期患者进行分类,并辅助改善组织学对肿瘤阶段的预测能力。以上研究表明利用生物信息学方法筛选癌症相关分子基因标志物的可行性,且筛选得到的基因具有较好的分类效果。
关于肿瘤发生的候选生物标志物筛选我们作了一些工作[9-10],也取得了比较好的结果。上述工作主要基于基因表达的差异度及其对分类的贡献大小来筛选,在此基础上发现一些生物学功能明确且生物学过程清楚的分子标志物并建立预测模型是本文的重点。通过统计学并结合基因的生物学通路分析筛选早期LUSC的分子标志物的方法,为肿瘤标志物的筛选提供了新的视角,筛选得到的标志物可能成为LUSC的诊断及治疗靶点,有助于LUSC的分子机制的研究,建立的肿瘤预测模型能够提高LUSC的与预测准确率,为科学研究和临床诊断提供了一种新途径。
1 数据与预处理
1.1 数据
1.1.1 TCGA data
训练集数据来源于癌症基因组图谱(The Cancer Genome Atlas, TCGA)公共数据库,下载的数据包括癌症基因表达谱数据和临床信息数据。使用Python语言编写程序将病人的表达谱和临床信息进行整合,挑选出正常组织和癌症I期的样本表达谱数据,同时为了便于后续的研究,删去在所有样本中表达值为空的基因。
最终收集到肺鳞状细胞癌样本295例(LUSC,49正常人,245癌症I期)、甲状腺癌样本342例(THCA,56正常人,286癌症I期)、肝细胞癌样本222例(LIHC,49正常人,173癌症I期)和肾透明细胞癌样本345例(KIRC,72正常人,272癌症I期)。
1.1.2 GEO data
从GEO数据库下载肺癌的基因表达数据(数据集为GSE11969),作为独立测试集对模型进行检验。仅保留信息完整的LUSC肿瘤I期和正常肺组织样本的基因表达数据,共得到样本数22例,其中癌症I期样本有17例,癌旁组织样本5例。
1.2 预处理
由于基因的表达数据相差较大,为了方便后期建模且训练收敛迅速,需要对基因表达谱数据进行归一化处理,要求其区间为[0,1],取值公式为:
(1)
其中:xmin为该基因在所有样本表达值的最小值;xmax为该基因在所有样本表达值的最大值。
2 方 法
2.1 肺鳞状细胞癌早期特征基因的筛选方法
本文涉及的分类问题为常见的二分类模型,但是由于特征的维度远远大于样本数,特征之间的关联关系相对复杂、关联关系间依赖性影响等问题,使得学习产生了诸多问题,比如:分析数据、训练模型时间长,数据量大导致“维度灾难”,使得模型过于复杂等等。为了克服这些不利因素的影响,提高特征识别的准确率,需要对数据集的特征基因进行筛选。本文在运用统计学的基础上,结合基因的生物学功能,建立一套分子标志物的筛选流程。
(1) 相关性筛选 相关性筛选可以计算出两个指标之间的相关系数,相关系数越大代表两个指标信息之间的相关性越强。在这里,两个指标分别代表着基因的表达值于癌症分期(正常肺组织和癌症I期)的相关性。本文采用斯皮尔曼公式计算相关性,设定相关系数r,保留相关系数r大于0.5的基因作为候选基因。
(2) t检验筛选 t检验过程是对两个癌症分期(癌症I期和正常)均数差别的显著性进行检验。对于筛选出的特征基因,满足齐性分布的采用双总体t检验进行筛选,保留对癌症分期具有显著性差异的特征基因,不满足齐性分布的基因采用非参检验中的Cruskal-Wallis秩和检验进行筛选,同样保留对癌症分期具有显著性差异的特征基因。
(3) 置信区间筛选 置信区间是一种常用的区间估计方法,根据基因的表达在癌症I期和正常肺组织中的差异性筛选基因,通过置信区间是否重合筛选出一组具有明显差异表达的基因,作为肺鳞状细胞癌差异基因子集1。其中,置信区间的筛选i值设为1.0。
(4) 弹性网络筛选 弹性网络可以克服噪声和变量相关性的影响,原理是将分类贡献较小的自变量的系数降为零。并且,当特征之间存在较强的共线性时,弹性网络可以标记出所有的变量,从而形成一个特定的基因组合。运用弹性网络(α= 0.5)的方法迭代筛选出另一组最优肺鳞状细胞癌差异基因子集2。
(5) KEGG通路富集筛选 KEGG通路分析可以确定不同样本间差异基因所参与的最主要代谢途径和信号转导途径。通过对差异基因做通路分析和生物学功能分析可以辨认LUSC的重要通路及其重要基因。采用DAVID在线分析平台对上述的两组特征基因集(LUSC特征基因集1和LUSC特征基因集2)分别进行KEGG通路分析,找出两组特征基因参与的共同通路,并进一步筛选出通路内的共有基因,得到候选基因集。具体流程图如图1所示。
为了验证筛选出的基因具有足够高的癌症分类能力,我们需要对候选基因集进行模式识别模型预测。对候选基因集,采用支持向量机(Support Vector Machine, SVM)、随机森林(Random Forest , RF)、人工神经网络(Artificial Neural Network, ANN)多种分类模型进行建模预测,并利用10 折交叉验证法对模型参数进行优化。
用混淆矩阵作为模型的评判标准,混淆矩阵的概念图如图2所示。其中,Positive是肺鳞状细胞癌癌症I期样本,Negative是正常肺组织样本。由此可推出,TP是指实际上是癌症I期样本并且也推断是癌症I期样本的样本个数;TN是指实际上是正常肺组织样本,预测值也为正常肺组织样本的样本个数;FN是指实际上是癌症I期样本,但预测为正常肺组织样本的样本个数;FP是指将正常肺组织样本预测为癌症I期样本的样本个数。
通过计算总体准确率(Accuracy, ACC)以及敏感度(Sensitivity, SEN)、特异度(Specificity, SPE)、马修斯相关系数(Matthews Correlation Coefficient, MCC)指标来评价模型的分类结果。其中,敏感性指的是肺鳞状细胞癌癌症I期样本的分类准确率,特异性指的是正常肺组织样本的分类准确率,而马修斯相关系数指标考虑到了TP、FP、TN和FN,对于不平衡的数据集也可以衡量。
ACC,SEN,SPE与MCC的定义如下:
(2)
(3)
(4)

图1 肺鳞状细胞癌分子标志物的筛选流程图Fig.1 Flow chart of molecular markers identification for LUSC
图2 混淆矩阵概念图Fig.2 Confuse matrix concept diagrams
(5)
2.2 Fisher判别建模
Fisher判别分析是多元统计分析判别归属的一种方法,其基本思想是将高维数据点投影到低维空间(一条直线)上,找到一个投影轴使得样本投影到该空间后能在保证方差最小的情况下,将不同类的样本更好的分开。它能根据已有类别的若干样本的数据信息,总结出分类的规律性,建立判别公式和判别准则,当遇到新的样本时,能判定该样本所属的类别。
本节的目的是在筛选出的分子标志物的基础上,建立基于Fisher判别分析的分类预测模型。首先,以候选基因集中的每个基因作为单一变量建立模型;然后,对12个分子标志物进行两两配对建模;最后,对所有基因按照准确率的大小进行排列,依次迭代建模。以模型的准确率(Accuracy, ACC)以及敏感度(Sensitivity, SEN)、特异度(Specificity, SPE)、马修斯相关系数(Matthews Correlation Coefficient, MCC)作为指标来评价模型的分类结果,通过多个模型的对比,选择表现最好的模型作为最终的分类预测模型。
3 结果与讨论
3.1 分子标志物的提取结果与讨论
3.1.1 分子标志物提取结果
在运用统计学方法的基础上综合分析基因的生物学功能,通过差异基因在通路上的富集,筛选得到3条共有通路和12个共有基因,如表1所示,筛选出的共有基因作为候选分子标志物组成候选基因集。
ROC曲线可以筛查出对癌症的识别能力较高的基因,曲线下的面积越大,说明该基因的诊断效能越大,故保留ROC曲线下面积大于0.9的基因。对所有的候选分子标志物进行ROC曲线分析,发现所有候选分子标志物的AUC 的值都在0.98以上,远远大于0.9,说明候选基因对癌症的诊断有意义,所以最终保留候选基因集中的12个基因作为分子标志物,分别是CLDN18,CD34,ESAM,JAM2,CDH5,F11,F8,CFD,MRC1,MARCO,SFTPA2,SFTPA1。

表1 肺鳞状细胞癌的KEGG通路分析结果Table 1 KEGG pathway analysis of LUSC
3.1.2. 相关通路及分子标志物的功能性分析
如前所述,经过KEGG通路分析后共富集到3条共有通路,分别是细胞粘附分子通路、补体通路和吞噬体通路,相应基因标志物在通路中的相关位置用红色星星加以标注。筛选得到LUSC的分子标志物共有12个,分别是CLDN18,CD34,ESAM,JAM2,CDH5,F11,F8,CFD,MRC1,MARCO,SFTPA2,SFTPA1。
图3是分子标志物在LUSC癌症I期和正常组织中的基因表达水平,由图可以看出,这12个基因在癌症Ⅰ期中表达量都低于正常组织组织中的表达量,说明这些基因的表达在LUSC早期的发生发展中受到了抑制。
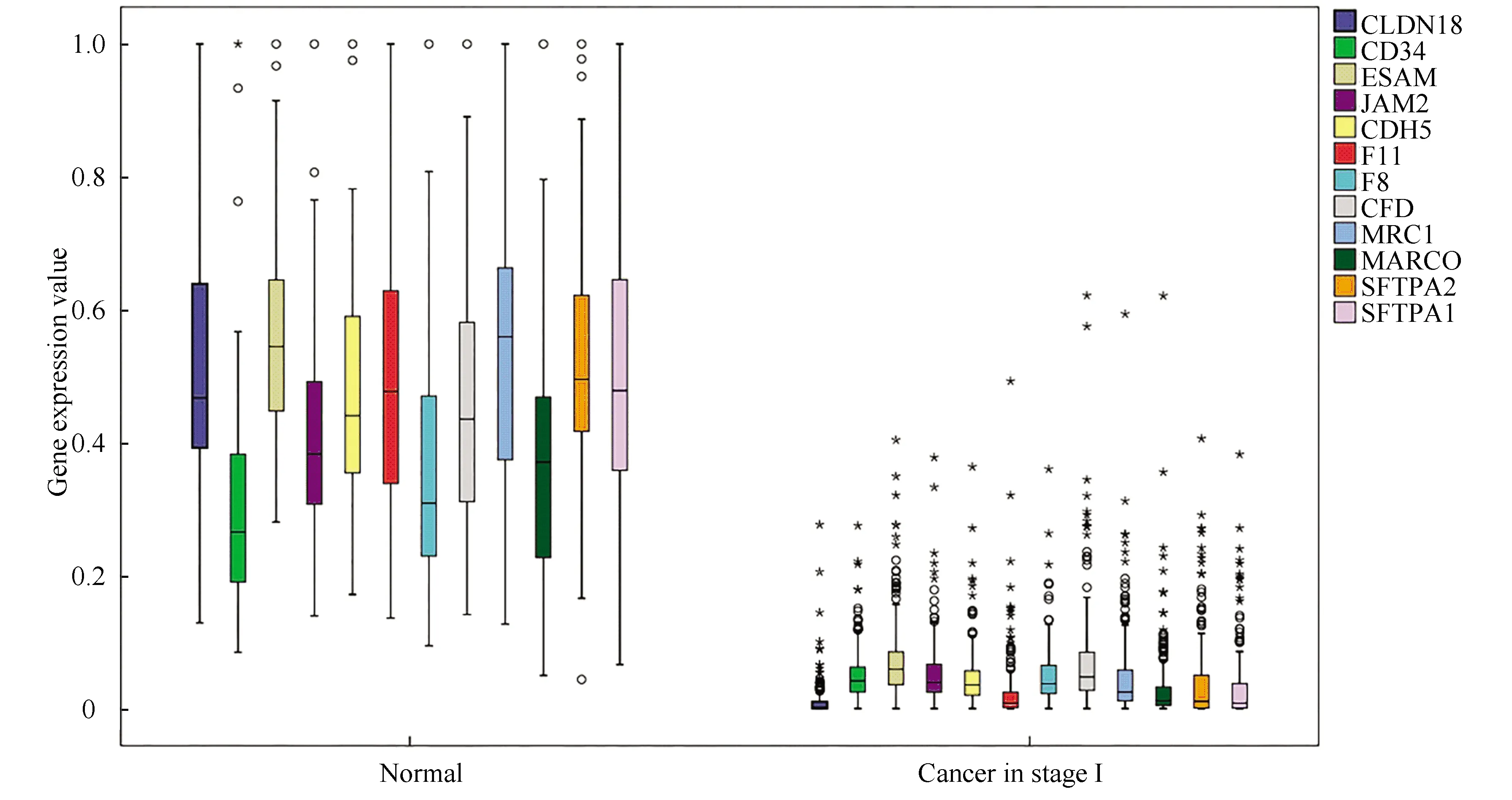
图3 在肺鳞状细胞癌癌旁组织和癌症I期中基因表达水平箱线图Fig.3 Box chart of gene expression levels in normal tissue and stage I cancer
(1)细胞粘附分子通路图如图4所示,12个基因标志物中的CLDN18、CD34、ESAM、JAM2、CDH5等基因属于该通路且处于重要位置,这些基因编码的蛋白作为细胞粘附分子在通路中发挥作用。细胞黏附分子是众多介导细胞间或细胞与细胞外基质间相互接触和结合的膜表面糖蛋白分子的统称,以受体-配体结合的形式发挥作用,使细胞与细胞间发生粘附,细胞间黏附作用密切参与体内免疫应答、炎症发生、凝血、肿瘤转移以及创伤愈合等一系列重要生理病理过程。由图2可知,基因CLDN18、CD34、ESAM、JAM2、CDH5在癌组织中的表达下调,这会导致细胞膜的通透性增加,为肿瘤细胞的侵袭和转移提供了机会。CLDN18编码的蛋白质位于上皮细胞和内皮细胞,是紧密连接的重要结构成分[11],紧密连接结构的丧失可导致肿瘤细胞的侵袭和转移[12]。有文献报道,CLDN18与胃癌的发生有关[13-14]。CD34编码的Ⅰ型跨膜糖蛋白作为一种黏附分子,在乳腺肿瘤中出现了异常表达,与乳腺肿瘤从良性到恶性发展过程中起重要作用[15]。ESAM表达于内皮细胞,在血管紧密连接中含量丰富,该基因的缺失可使内皮细胞的通透性增加,即ESAM的表达同细胞通透性呈负相关,表明该基因与肿瘤细胞的侵袭和转移有重要联系[16]。JAM2在上皮细胞和内皮细胞的细胞连接处以及红细胞、白细胞和血小板的表面富集,Kok-Sin等[17]发现JAM2基因在结直肠癌中高甲基化、低表达,与肿瘤的发生发展有关。CDH5编码的蛋白在内皮细胞粘附连接的组装和维持中发挥作用,CDH5在多种肿瘤中异常表达,包括侵袭性乳腺癌、非转移性肾细胞癌、侵袭性黑色素瘤和骨肉瘤等,与癌症的发生发展密切相关[18,21]。
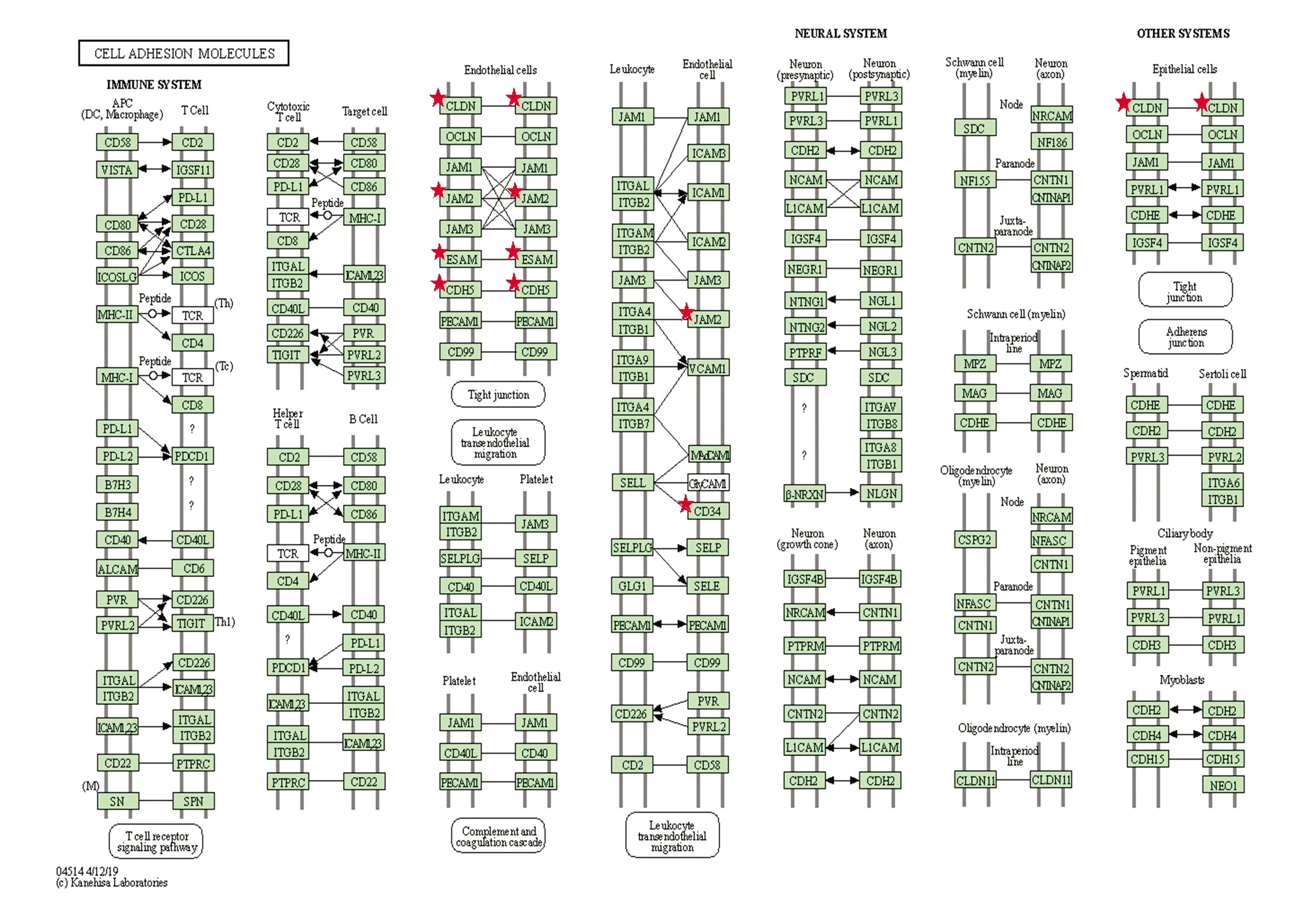
图4 CLDN18、CD34、ESAM、JAM2 和 CDH5在细胞粘附分子中的位置Fig.4 Positions of CLDN18,CD34,ESAM,JAM2, and CDH5 in cell adhesion molecules
(2)补体系统通路图如图5所示,基因F11、F8和CFD在该通路中处于重要位置。补体系统又分为两条通路,分别是凝血级联和补体级联,它们相互独立又相互联系。其中,凝血级联是机体形成血块以防止失血的过程,F11和F8编码的蛋白是凝血级联中的凝血因子,参与了凝血的内在途径,在肿瘤的微环境中,微小血栓的形成会限制血液供应从而抑制肿瘤细胞的生长,进而限制肿瘤细胞向周围组织浸润及转移[22];补体级联中起重要作用的是补体,补体是一种存在于血清、组织液和细胞膜表面的一组经活化后具有酶活性的蛋白质,对机体的防御功能、免疫系统功能的调节以及免疫病理过程都发挥重要作用,CFD编码的补体因子存在于补体系统的旁路途径中,具有级联放大作用,在文献[23]中发现,肝细胞癌患者的血清补体含量低于健康样本,可能的原因为肿瘤细胞表达补体的抑制剂,从而抑制补体的激活,阻止了补体的级联放大效应并大量消耗补体,导致肿瘤对机体免疫监督的逃逸,促进了肿瘤的发生与进展[24]。
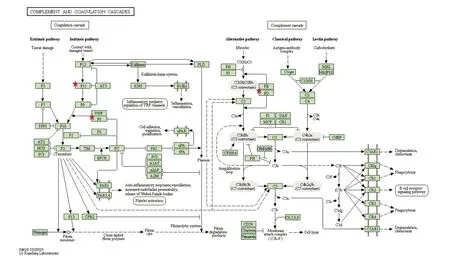
图5 F11、F8和 CFD 在补体通路中的位置Fig.5 Positions of F11,F8, and CFD in complement and coagulation cascades
(3) 吞噬通路图如图6所示,基因MRC1、MARCO、SFTPA2和SFTPA1在该通路中发挥着重要作用。吞噬体是巨噬细胞参与组织重塑、清除凋亡细胞、抑制细胞内病原体扩散的天然能力的关键细胞器,是一种在免疫过程中常见的细胞结构。巨噬细胞的吞噬作用是细胞内化外来的大颗粒物质的过程,在先天免疫和适应性免疫中非常重要,有助于我们对抗传染病的能力。巨噬细胞在不同的环境下会发生不同性质的活化,而活化后的巨噬细胞具有不同的免疫功能。通常巨噬细胞有3种活化形式,分别是经典活化的巨噬细胞(M1型巨噬细胞)、替代性活化的巨噬细胞(M2型巨噬细胞)和II型活化的巨噬细胞。其中,M1型和M2型巨噬细胞与肿瘤的发生、发展和转移密切相关,M1型巨噬细胞能够分泌一氧化氮(NO)等杀伤分子,可以促进炎症的发展,加速感染的病原体和肿瘤细胞的凋亡;而M2型巨噬细胞被诱导剂活化后,使得甘露糖受体和清道夫受体表达上调, 减少NO的分泌, 从而降低了杀伤细胞内病原体的能力,对肿瘤细胞的增值和侵袭起促进作用[25]。基因MRC1编码的甘露糖受体和MARCO编码的清道夫受体是肺泡巨噬细胞的膜受体,其在早期LUSC中的低表达可能预示着肺泡巨噬细胞活化为M1型巨噬细胞,从而对肿瘤细胞的发展起抑制作用,并在抗原识别、内毒素清除、细胞粘附和调控炎性递质分泌等机体防御反应中起重要作用[26]。SFTPA1和SFTPA2 是两个同源基因,属C型胶凝素超家族(Collectins)成员,由肺泡II型上皮细胞分泌,作为肺内原始的内源性的免疫调节剂,具有维持肺部稳定、调节局部免疫和炎症反应以及参与局部防御等功能[27-28],且由于SFTPA具有肺部特异性,因此其有望用于肺部疾病的治疗及作为新的肺疾病生物学标志物[29]。MRC1和MARCO作为肺泡巨噬细胞表面的模式识别受体,可以识别病原体从而引发吞噬运动,而SFTPA1和SFTPA2编码的SFTPA能与许多不同的病原体包括病毒、真菌、细菌结合,加速它们被肺泡巨噬细胞吞噬和消灭,这在肺的原发性防御免疫系统中发挥着重要作用。
综上所述,通过对分子标志物的相关通路以及基因功能性分析,发现CLDN18,CD34,ESAM,JAM2,CDH5,F11,F8,CFD,MRC1,MARCO,SFTPA2和SFTPA1这12个分子标志物与LUSC的发生发展密切相关,已有文献也验证了其在癌症中的重要性,这也表明本文的筛选方法是可靠的。
3.1.3. 评估LUSC的分子标志物
(1)对LUSC 和正常肺组织的识别能力 目的是识别对肺鳞状细胞癌早期的发生有着至关重要的分子标志物。为进一步揭示LUSC发生和发展的机理奠定基础,并对LUSC的早期诊断提供理论支持,因此识别的基因必须具有足够高的癌症分类能力,采用的分类模型必须具有高的分类精度。对本文方法筛选得到的分子标志物分别采用支持向量机(SVM)、随机森林(RF)、人工神经网络(ANN)三种分类模型进行建模,分类结果如表2所示。

图6 MRC1、MARCO、SFTPA2 和 SFTPA1在吞噬通路中的位置Fig.6 Positions of MRC1,MARCO,SFTPA2, and SFTPA1 in phagosome

表2 肺鳞状细胞癌分子标志物的模式识别结果Table 2 Pattern recognition results of LUSC molecular markers
从表中可以看出,对于三种分类模型分类结果,模型SVM和模型ANN的敏感性达到1,说明模型对于癌症I期的识别准确率最高;模型RF的敏感性达到了0.99,说明其对癌症I期也具有很高的识别准确率。对三种分类模型的结果综合对比可以看出,三种模型的准确率都达到了99%左右,马修斯相关系数的值也都达到了0.96以上,表明筛选出的分子标志物对肺鳞状细胞癌癌旁和I期的分类都具有很高的区分能力,也说明了通过通路富集和基因的生物学功能分析筛选分子标志物的方法具有有效性和可靠性。
(2)对LUSC 和其余癌症的识别能力 为了验证LUSC分子标志物的特异性,即区分LUSC 和其他癌症的能力,使用支持向量机建立肿瘤分类模型。表3显示了12个LUSC的分子标志物对于LUSC样本和甲状腺癌样本、肝癌样本、肾透明细胞癌样本的模式识别结果。

表3 肺鳞状细胞癌的分子标志物区别肺鳞状细胞癌与其他癌症的结果Table 3 Results of LUSC molecular markers distinguishing between LUSC and other cancers
由表3可知,LUSC分子标志物对LUSC的识别度很高,敏感性达到96%以上,在排除甲状腺癌时达到91.61%的特异性,排除肝癌和肾透明细胞癌的特异性也达到73%以上,表明使用12个LUSC分子标志物既能够对LUSC样本有极高的识别度,同时也能排除其他癌症样本,验证了本文的筛选方法得到的LUSC分子标志物具有特异性。
3.2 Fisher判别建立模型
为了建立肺鳞状细胞癌早期的分类预测最优模型,为临床早期预测及诊断提供一种新的辅助方法,我们需要对筛选出的分子标志物进行多种组合的Fisher判别分析建模,建立模型的基因尽可能少,并且模型必须具有足够高的癌症分类精度。
3.2.1. 单一标志物的判别模型分类结果
对候选基因集中的每个基因都进行建模预测,分类结果见表4。

表4 单一标志物的判别模型分类结果Table 4 Classification results of discriminant model for single marker
由表4中可以看出,在所有的模型中,敏感性都在0.98左右,说明其对于癌症I期的样本具有较好的识别能力;特异性在0.77以上,说明其对癌旁样本的识别能力有些不足。综合来看,建立的12个模型的准确率都在93%以上,其中基因SFTPA1和基因ESAM建立的模型准确率最高,都在98%以上,说明其模型对肺鳞状细胞癌癌旁和癌症I期具有很高的区分能力。
3.2.2 两个特征基因组合判别模型的分类结果
对12个特征基因进行两两配对建立模型,利用Fisher判别分析共建立了66个判别模型,取准确率及马修斯相关系数排名前十的判别模型,其预测结果见表5。
由表5可以明显看出,与单基因建立的模型相比, 两个特征基因组合的模型预测准确率更高,其诊断价值也更高。在这十个模型中,有7个模型的准确率达到了0.99。其中,前六个模型的特异性,即识别正常肺组织样本的能力达到了1,而敏感性(识别LUSC癌症I期样本)也达到了0.99,说明模型对肺鳞状细胞癌癌旁和癌症I期样本都具有很高的识别能力。

表5 两个特征基因组合模型的分类结果Table 5 Classification results of discriminant model for two characteristic genes
3.2.3 多个基因组合模型的诊断价值评估
将12个基因按照准确率的大小,从大到小排列,结果为SFTPA1、ESAM、CLDN18、CDH5、JAM2、SFTPA2、F11、MRC1、MARCO、CD34、F8、CFD。采用Fisher判别依次累加基因建立模型,模型预测准确率如图7所示。

图7 ACC与特征基因集大小的关系Fig.7 Relationship between ACC and size of characteristic genes set
由图7可以看出,当基因数量增加为2个时,LUSC的判别模型的分类准确率达到最高,此后当基因再次增加时,其模型的分类预测准确率不在有变化,一直保持稳定。
3.2.4 最优模型及其独立数据集验证
Fisher判别模型由判别函数来表示,通常采用重心距离来定义类与类之间的距离,从而对待判样本进行判别。通过对上述涉及的多个模型的分类结果对比,最终选择了基因SFTPA1和ESAM,以及以这两个基因建立的三个分类准确率较高的LUSC判别模型,分别是SFTPA1单基因模型、ESAM单基因模型以及SFTPA1和ESAM双基因模型。表6显示了三个模型对正常肺组织和LUSC癌症I期样本的判别重心以及模型的判别函数。综合考虑模型对LUSC的分类敏感性和特异性,最终保留SFTPA1和ESAM建立的Fisher判别模型。
为了验证判别模型的有效性,需要在不同的平台上下载LUSC的数据集作为验证集,验证模型对于LUSC的癌症分类能力。从GEO数据库中下载的GSE11969数据集,经过预处理之后,作为独立验证集参与模型的检验。SFTPA1和ESAM双基因判别模型对独立验证集的分类结果如表7所示。

表6 判别模型Table 6 Discriminating model

表7 独立验证集的分类结果Table 7 Classification result of independent verification set
独立验证集的分类结果显示,LUSC的判别模型的分类特异性为1,敏感性为0.88,说明该模型对LUSC癌症样本的识别能力相对较弱,但是对正常样本的识别能力非常好。总体准确率在90%以上,说明该模型对LUSC有较高的分类能力,且普适性较好。
4 结 论
通过基因组百科全书(KEGG)通路富集分析和基因生物学功能分析,结合统计学的相关方法筛选出12个LUSC发生的早期标志物,对这些基因采用机器学习的方法建模,得到准确率在98%以上,说明该方法筛选得到的分子标志物对LUSC早期癌症样本和正常组织样本的分类能力很好,且筛选得到的12个分子标志物(CLDN18,CD34,ESAM,JAM2,CDH5,F11,F8,CFD,MRC1,MARCO,SFTPA2,SFTPA1)可能成为LUSC的诊断及治疗靶点,有助于LUSC分子机制的研究。其次,本文建立了一种基于早期标志基因的肿瘤预测模型,对于不同平台的LUSC数据集,其分类准确率高于90%,说明该模型的普适性较好,具有良好的临床应用前景。虽然本文的研究具有良好的结果,但是研究内容还是仅仅停留在生物信息学预测层面,缺少实验验证和分析,后续应与临床试验合作开展检验工作,使得结论更加严谨。
致谢:本研究承蒙国家自然科学基金(No.11572014),国家科技部的重点研发项目(No.2017YFC0111104)以及智能化生理测量与临床转化北京国际科技合作基地的资助。
猜你喜欢
好日子(2021年8期)2021-11-04
健康之家(2021年19期)2021-05-23
医学食疗与健康(2021年27期)2021-05-13
农业科技与信息(2021年2期)2021-03-27
中国交通信息化(2018年5期)2018-08-21
海峡姐妹(2018年7期)2018-07-27
特别健康(2018年4期)2018-07-03
特别健康(2018年2期)2018-06-29
中华老年多器官疾病杂志(2016年9期)2016-04-28
医学研究杂志(2015年7期)2015-06-22
