
肺腺癌相关长链非编码RNA生物标志物识别
2021-01-04 09:58于晓庆
应用技术学报 2020年4期
于晓庆
(上海应用技术大学 理学院,上海 201418)
肺癌是全球因癌症导致人类死亡的最大因素。到目前为止,肺癌的5年生存率仅为18%[1]。在肺癌的亚型中,非小细胞肺癌约占85%。而肺腺癌(lung adenocarcinoma,LUAD)是非小细胞肺癌中最常见的恶性病理类型。近些年,随着靶向药物的出现,肺腺癌的5年生存率不断在提高,但由于缺乏有效的诊断和预后生物标志物,死亡率仍然很高[2-3]。 因此,为了降低肺腺癌患者的死亡率,迫切需要寻找有效的分子标志物,为患者的诊断和临床治疗提供一定的理论依据。
长非编码RNA(long noncoding RNA,LncRNA)是长度超过200个核苷酸的非编码基因。过去很长一段时间,这些非编码基因曾被认为是基因组的“暗物质”,并不具有生物学功能。然而,随着实验技术和计算机技术的发展,越来越多的研究表明,LncRNA通过不同的机制参与了细胞的几乎整个生命过程,并在细胞分化、代谢、细胞增殖和凋亡、转录、染色质水平的表观遗传学状态调控等许多基本和关键的生物过程中都起到了非常重要的作用[4-5]。同时,很多研究发现LncRNA与包括肺腺癌在内的多种癌症的发展和生存都有关联性,强调了LncRNA在肺腺癌发生和发展中的分子机制和生物学特性,表明某些lncRNA可以作为肺腺癌患者预后的分子标志物[6-7]。
个体化医疗的一个关键问题就是阐明疾病个体的分子机制。复杂疾病的发生,通常并不是由于单个分子的功能障碍导致,而是由于随着时间和条件的动态变化,相关系统或者网络的功能紊乱而引起[8-9]。一个样本个体在特定状态下的分子网络,称其为单样本网络。单样本网络是能够准确描述疾病个体在特定条件下疾病状态的关键个性化特征。对样本个体在特定条件下的单样本网络进行评估和分析,能够从系统和网络水平揭示复杂疾病发生发展的分子机制。本文基于TCGA数据库中肺腺癌的RNA-seq数据和临床数据,利用单样本网络方法构建肺腺癌患者的单样本网络模型,分析和识别出6个与肺腺癌相关的lncRNA分子标志物。功能富集分析表明,6个lncRNA可能参与了肺腺癌患者细胞的代谢过程。本文的研究可以为肺腺癌的诊断、临床治疗以及预后提供可能的分子标志物。
1 数据准备
从TCGA (The Cancer Genome Atlas) 数据库下载了肺腺癌的 RNA-seq 数据和临床试验数据,包括535个肺腺癌的组织样本(tumor samples)以及59个癌旁的组织样本(normal samples)。
2 方法
2.1 差异表达lncRNA的识别
我们使用R软件包“edgeR”对肺腺癌表达数据中所有的lncRNA进行基因的差异表达分析。该软件包为多组实验提供了精确的统计方法,它的一个特殊功能是经验贝叶斯方法,该方法即使在生物复制水平最低的情况下也可以估算基因特异性的生物变异。在我们的研究中,FDR<0.05且 |FC|>2为筛选差异表达的lncRNAs的临界条件。
2.2 单样本网络的构建
单样本网络是基于单个样本对于给定的一组参考样本的扰动分析而构建的,它可以精确地刻画个体样本的疾病状态[10]。单样本网络的构建需要两组数据,一组是参照样本数据,另一组是测试样本数据。通常,这两组数据有着共同的特征属性。在本研究中,59个肺腺癌的癌旁样本作为参照样本组,535个肺腺癌的肿瘤样本作为测试样本组。首先,利用皮尔森相关系数(pearson correlation coefficient,PCC)计算每对lncRNA基因表达谱之间的相关性从而得到一个相关性网络,称为参照网络(reference network),记为Nr。然后,再将一个测试样本s添加到该参考样本组中,利用上述相关性的计算方法,可以得到一个新的相关性网络,称为扰动网络(perturbed network),记为Np。最后,通过比较参照网络和扰动网络之间的差异获得一个差异网络,被称为样本s的单样本网络(single-sample network),记为Nssn(Nssn=|Nr-Np|)(见图1)。
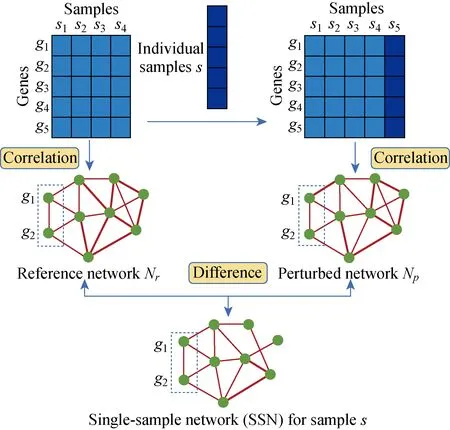
图1 单样本网络构建流程图Fig.1 Flowchart for constructing a single-sample network
按照上述方法,轮流将535个肿瘤样本依次作为测试样本添加到参照样本组中,可以得到535个单样本网络。容易看出,如果测试样本在基因表达模式上与参照样本组的表达模式相似,即使将测试样本添加到参照样本组中,任何2个分子之间的PCC变化将非常小。相反,如果测试样本和参照样本组在表达模式上存在明显差异,则将测试样本添加到参照样本组后将引起明显的变化。可以看出,单样本网络是由样本s决定的,因此它可以用来作为描述样本s的一个特定特征。我们对所有的肿瘤样本构建其对应的单样本网络并进行统计分析,筛选出可能与肺腺癌相关的lncRNA分子标志物。
2.3 生存分析
COX比例风险回归模型,是由英国统计学家D.R.Cox 提出的一种半参数回归模型。该模型以生存结局和生存时间为因变量,能够分析各种影响因素对病人生存期的影响。COX回归分析是医学领域对于疾病预后分析最常用的统计方法之一。我们从TCGA数据库下载了肺腺癌病人的临床试验数据,同时筛选掉没有临床数据的样本,一共得到512个肺腺癌的样本。利用单变量Cox回归分析来评估生存率和每个lncRNA表达水平之间的关联性。
2.4 功能分析
到目前为止,对与肺腺癌相关的lncRNA的功能机制的报道还很少。为了进一步验证与肺腺癌相关的lncRNA的分子功能,利用相关性分析计算了并识别出lncRNA的共表达基因,通过对其共表达基因功能的探索,进而研究lncRNA的分子功能。这里,把计算结果中PCC>0.45,且P<0.01的编码基因,认为是识别出的lncRNA的共表达基因。
Metascape(http://metascape.org/gp/index.html)是一个免费的基因注释和分析资源,可以帮助研究者了解一个或多个基因的相关功能[11]。我们通过Metascape对共表达基因进行Go和KEGG功能富集分析。这里,P的临界值设定为P<0.01。
3 结果与讨论
3.1 差异表达分析结果
从下载的RNA-Seq数据中,我们提取了 14 823 个lncRNA的表达数据。 其中,肿瘤样本未表达数量超过10% 的lncRNA数据被剔除,共得到了 4 311 个lncRNA。通过edgeR差异表达分析得到了 1 178 个差异表达的lncRNA,其中包括851个表达下调的lncRNA和327个表达上调的lncRNA(见图2)。图2中3个部分分别表示表达下调、无差异表达、表达上调的lncRNA。
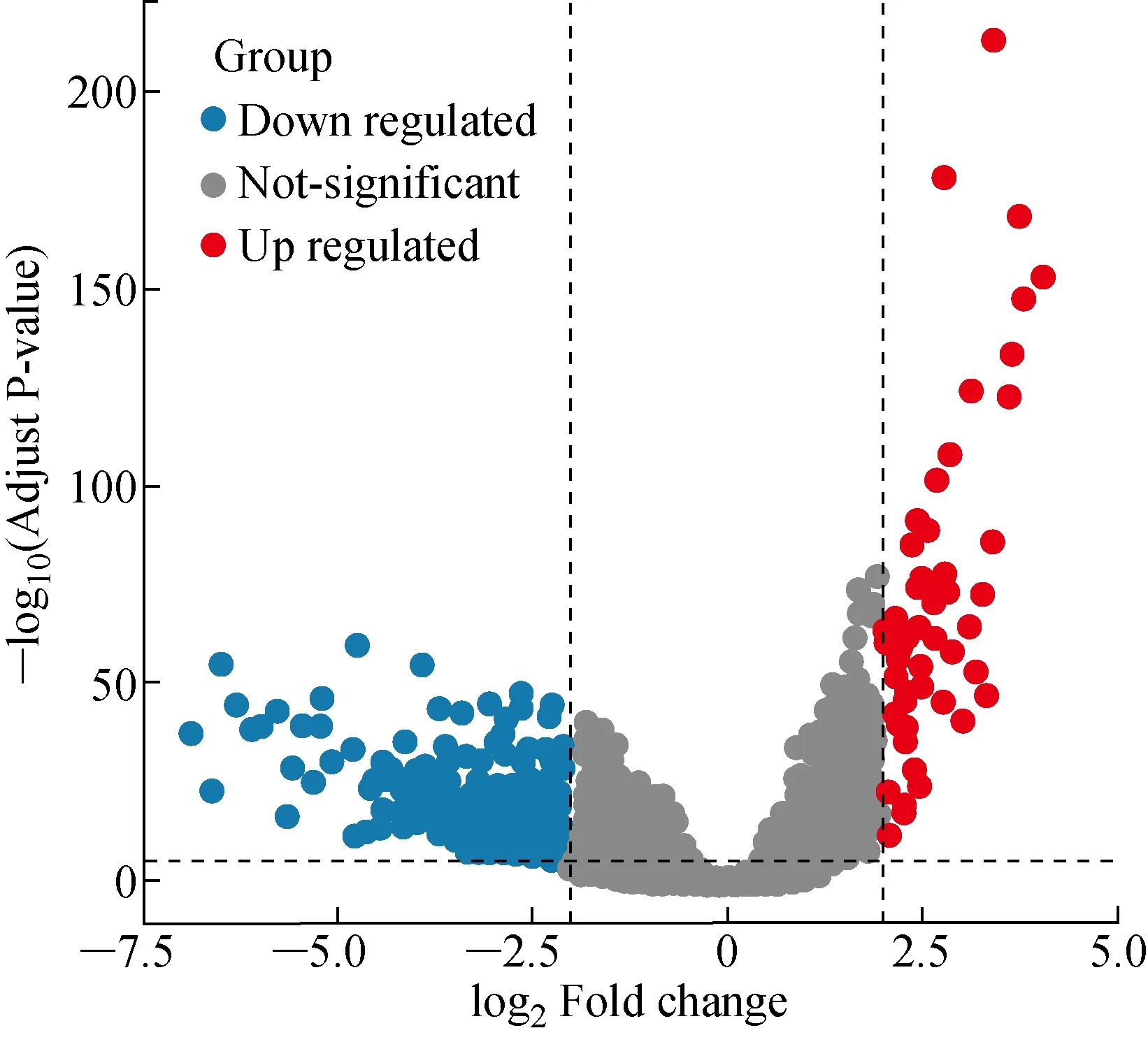
图2 差异表达lncRNAs的火山图Fig.2 The volcano plot of the differentially expressed lncRNAs
3.2 单样本网络分析结果
为了方便计算,我们把通过2.2得到的单样本网络转化为一个邻接矩阵 ΔD。容易知道,ΔDi,j表示单样本网络中第i个lncRNA和第j个lncRNA在参照网络和扰动网络中对应边的PCC的差值,记为ΔPCC。基于假设:如果某个lncRNA可能是肺腺癌相关的分子标志物,则在差异网络中,由该lncRNA连接的所有边对应的ΔPCC的总和将比其他lncRNA对应的总和更高。这里,用向量SD来表示 1 178 个lncRNA在每个单样本网络中连接的所有边所对应的ΔPCC的总和,记作
SD=(SD1,SD2,…,SDi,…,SD1 178)T
(1)
式中,SDi可通过以下方式计算:
(i=1,2,…,1 178,j=1,2,…,1 178)
(2)
为了找到与肺腺癌相关的lncRNA分子标志物,我们对每个单样本网络中所有lncRNA的SDi值进行排序。SDi的值越大,其成为肺腺癌的分子标志物的可能性也就越大。对于所有535个单样本网络,我们用一个矩阵M1 178×535来表示
M=
i=1,2,…,1 178,j=1,2,…,535
(3)
式中,SD1,j>SD2,j>…>SDi,j>SD1 178,j,i表示第i个lncRNA,j表示第j个肿瘤样本。
我们取上述矩阵M的前10行(即在每个肿瘤样本排序中的前10个lncRNA)进行统计分析。在535个肿瘤样本中,出现频率最高的30个lncRNA作为我们通过单样本网络方法识别出的可能与肺腺癌相关的候选lncRNA分子标志物。
3.3 生存分析结果
从肺腺癌患者的临床数据中选择整体生存期(overall survival,OS)进行单因素cox风险分析,结果显示在30个lncRNA中有6个与患者的OS显著相关(P<0.05)。6个lncRNA的详细信息见表1。基于这6个lncRNA建立了一个风险得分评估模型,式为:OS-risk score=0.034×LINC00460+0.057×RP11-284F21.9+0.426×RP11-539E17.5+0.227×CTD-2377D24.6+0.053×RP1-27K12.4+0.007×RP11-783K16.5。式中的6个系数分别为6个lncRNA在生存分析回归模型中对应的风险系数,基因名代表该lncRNA的表达数据。利用上述公式对512个肺腺癌患者进行风险得分计算,并根据所有患者风险得分的中位数将患者分为高低风险两组。这512个肺腺癌患者在预后模型中的分布以及生存时间的分布见图3(a)。ROC曲线被用来评估该风险得分评估模型的预测性能,结果显示该模型在生存时间为1年、3年和5年时间的AUC值分别为0.68、0.7和0.71,这说明在5年的生存期内,随着生存时间越来越长,该风险评估模型对于患者生存预后的准确性也越来越好,见图3(b)。同时也采用KM生存曲线对这些患者的生存情况进行了分析,结果显示低风险组患者的死亡率显著低于高风险组(HR=2.72,95%CI=2.13~3.48,P<0.000 1),见图3(c)。
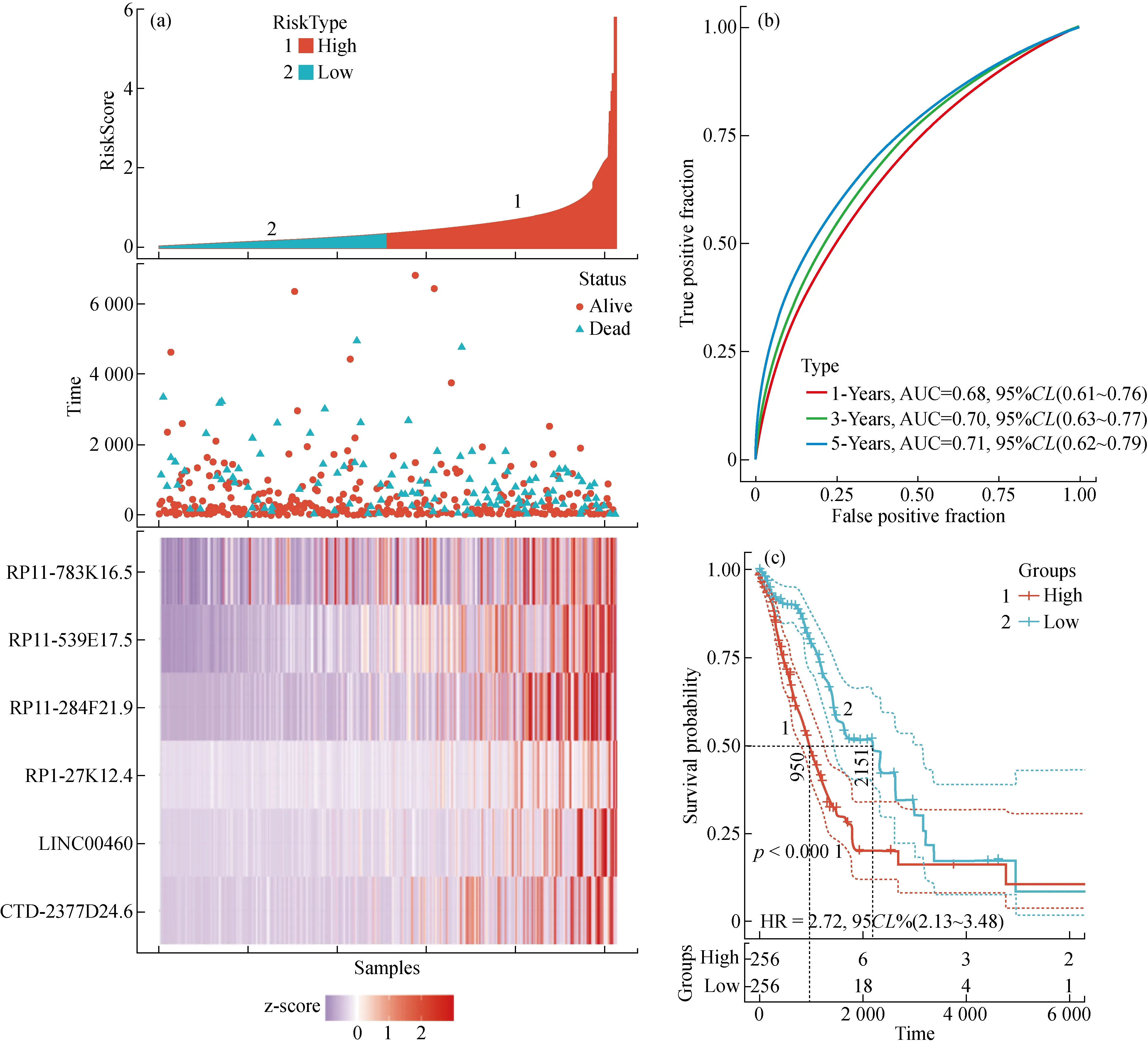
图3 基于6个lncRNA的风险得分对肺腺癌患者进行预测的结果(a)风险得分分布情况和肺腺癌患者的生存状态;(b)整体生存情况预测的风险得分ROC曲线分析;(c)高风险组(n=256)和低风险组(n=256)患者整体生存情况的KM分析Fig.3 The result of predicting the overall survival of patients with LUAD based on the risk score of the 6 lncRNAs (a)The risk score distribution and patients’ survival status,(b)The receiver operating characteristic (ROC) analysis of the risk score for predicting the overall survival,(c)Kaplan-Meier analysis of patients’ overall survival in the high risk (n=256) and low-risk (n=256)

表1 与肺腺癌具有显著预后关系的6个lncRNATab.1 The 6 lncRNAs significantly associated with prognosis of LUAD patients
3.4 功能富集分析结果
在共表达基因分析中,与6个lncRNA至少1个相关的蛋白质编码基因就被认为是显著相关的共表达基因。通过皮尔森相关分析计算,一共得到了52个共表达基因,这些基因主要与LINC00460、RP11-284F21.9、RP11-539E17.5、CTD-2377D24.6、RP11-783K16.5这5个lncRNA相关,见表2。采用Metascape对52个共表达基因进行Go和KEGG功能富集分析,见图4。图4中每个条形代表富集到的一簇功能,可以看到共表达基因主要与辅因子代谢过程、类固醇代谢过程以及细胞酮等代谢过程有关,而这些代谢过程是疾病产生和发展过程中的重要影响因素。

表2 5个lncRNA的共表达基因Tab.2 The co-express genes of the 5 LncRNAs

图4 共表达基因功能富集分析(a)共表达基因功能富集热图;(b)共表达基因富集GO的详细网络结构Fig.4 Functional enrichment analysis of the co-express genes (a) Heatmap of the functional enrichment analysis of the co-expressed genes,(b)Detailed net structure of enriched GO term of the co-expressed genes
4 结 语
基于TCGA数据库中肺腺癌的表达数据,利用基因差异分析,单样本网络方法以及生存分析识别出与肺腺癌相关的6个lncRNA生物标志物。利用单样本网络识别lncRNA 生物标志物,是对样本个体特定条件下动态分子网络进行分析研究,对于揭示复杂疾病在网络水平的LncRNA分子机制有着积极的指导意义,也为从系统水平识别人类复杂疾病相关LncRNA生物标志物提供了一个新视角。
猜你喜欢
安徽农业科学(2022年1期)2022-02-14
昆明医科大学学报(2020年12期)2021-01-26
中成药(2018年7期)2018-08-04
中华老年多器官疾病杂志(2016年9期)2016-04-28
中国学术期刊文摘(2016年1期)2016-02-13
中国学术期刊文摘(2016年1期)2016-02-13
西南军医(2016年3期)2016-01-23
医学研究杂志(2015年7期)2015-06-22
中国当代医药(2015年17期)2015-03-01
现代检验医学杂志(2015年6期)2015-02-06
