
黄芪药材提取纯化过程中黄芪皂苷Ⅳ含量变化及相关成分分析
2020-12-29 08:56吴惠勤黄晓兰刘梦云
分析测试学报 2020年12期
陈 超,吴惠勤,黄晓兰,黄 芳,刘梦云
(广东省科学院 广东省测试分析研究所(中国广州分析测试中心) 广东省中药质量安全 工程技术研究中心,广东 广州 510070)
黄芪为豆科植物蒙古黄芪Astragalusmembranaceus(Fisch.) Bge.var.mongholicus(Bge.) Hsiao 或膜荚黄芪Astragalusmembranaceus(Fisch.) Bge.的干燥根[1]。黄芪甲苷即黄芪皂苷Ⅳ,因具有抑制结肠癌细胞增殖、抗糖尿病、抗炎等多种药理活性[2-4],自1995年版《中华人民共和国药典》开始被作为黄芪药材质量控制的重要指标之一。
2015年版《中华人民共和国药典》[1](以下简称《中国药典》)中黄芪含量测定项下,样品采用索氏提取,提取液蒸干后用正丁醇萃取和氨试液除杂,正丁醇层蒸干后再利用D101大孔树脂纯化,纯化后的溶液蒸干后定容,利用高效液相色谱-蒸发光检测器(HPLC-ELSD)测定黄芪皂苷Ⅳ含量。与《中国药典》相比,《香港中药材标准》[5](以下简称《港标》)对黄芪皂苷Ⅳ的测定方法更为简便,采用甲醇超声提取,用氨试液溶解提取物后再用水饱和的正丁醇萃取,测得的黄芪皂苷Ⅳ含量与《中国药典》法测得的含量无显著性差异[6-7]。《中国药典》和《港标》对黄芪药材中黄芪皂苷Ⅳ含量测定的前处理过程中均使用氨试液,但碱性溶液在去除黄酮等酚酸类物质的同时,会引起其他皂苷类成分脱掉乙酰基或丙二酰基[8]而转化为黄芪皂苷Ⅳ,如黄芪皂苷Ⅰ和Ⅱ、异黄芪皂苷Ⅰ和Ⅱ、乙酰黄芪皂苷Ⅰ等[9];不同的碱处理方法和处理时间的长短均会影响黄芪皂苷Ⅳ的得率[10-11]。《中国药典》和《港标》测得的黄芪皂苷Ⅳ含量并不是黄芪药材中原始的含量,而是包含了其他黄芪皂苷类成分在碱性溶液中水解后的含量。由于氨试液振摇时间和振摇后放置时间均未明确规定,在实验过程中因检测人员操作的差异性,导致黄芪皂苷Ⅳ同系物水解不完全,使得黄芪皂苷Ⅳ的重复性不佳[12],影响了对药材进行准确的质量评价。
目前,关于黄芪药材中黄芪皂苷Ⅳ含量测定的方法主要有薄层扫描法、分光光度法、近红外光谱法和HPLC法,其中HPLC-ELSD法最为常见。黄艳伟等[13]采用加速溶剂萃取法(ASE)减少了溶剂用量和提取时间,王玉等[14]采用Box-Behnken设计-响应面法优化了黄芪药材提取工艺中的乙醇浓度、提取时间和提取次数,这些文献中关于黄芪皂苷Ⅳ的净化过程均采用《中国药典》方法。此外,有文献[15]采用在线固相萃取(SPE)-液相色谱法结合电雾式检测器(CAD),将黄芪药材用0.1 mol/L 氢氧化钾-甲醇提取后,直接用其特有的装置进行含量测定,自动化程度高,确保了黄芪皂苷Ⅳ的提取率和重现性,但使用SPE柱加大了检测成本。液相色谱-质谱(LC-MS)法具有高灵敏度和选择性,可以简化样品前处理过程,提高分析效率,适用于复杂样品中微量成分的检测。夏义平等[16]用碱性溶液提取药材,调至中性后直接采用超高效液相色谱-串联质谱(UPLC-MS/MS)法测定黄芪中黄芪皂苷Ⅳ的含量,测定结果包含了转化后的黄芪皂苷Ⅳ。
本文采用UPLC-MS/MS法考察了不同提取方式和净化操作对黄芪药材中游离黄芪皂苷Ⅳ提取率的影响,并利用具有超高分辨率的傅里叶变换离子回旋共振质谱仪(FT-ICR-MS)分析其相关成分的差异性。本研究还阐明了4种黄芪皂苷Ⅳ同系物之间的转化途径,确定了导致《中国药典》测定结果不稳定的原因,优化了含量测定的方法,最终建立了一种稳定、高效的黄芪药材中游离黄芪皂苷Ⅳ含量测定方法,可为准确评价黄芪药材质量提供重要依据。
1 实验部分
1.1 仪器与试剂
Agilent 1290 Infinity Ⅱ UPLC/6470 QQQ 超高效液相色谱-质谱联用仪(美国Agilent公司);Agilent 1290 Infinity Ⅱ/Solarix 7.0 T 傅里叶变换离子回旋共振质谱仪(美国Agilent公司/德国Bruker公司);AS 3120超声波发生器(天津奥特赛恩斯仪器有限公司);HH-2型恒温水浴锅(常州澳华仪器有限公司);FCE-3000索氏提取器(上海乔跃电子有限公司);赛多利斯TP-114电子天平(美国Sartorious公司);D101型大孔吸附树脂(东鸿化工有限公司);黄芪甲苷(黄芪皂苷Ⅳ)对照品(中国食品药品检定研究院,批号:110781-201717,含量:96.9%);乙腈(色谱纯,德国Merck公司);甲酸(LC-MS级,美国Thermo Fisher Scientific公司);实验用水为二次蒸馏水,其余试剂均为分析纯。
实验所用黄芪药材(Y1、Y2和Y3)均为客户委托送检样品,产地为甘肃,经专家鉴定为蒙古黄芪Astragalusmembranaceus(Fisch.) Bge.var.mongholicus(Bge.) Hsiao(Y1和Y2)或膜荚黄芪Astragalusmembranaceus(Fisch.) Bge.(Y3)的干燥根。
1.2 标准溶液的配制
精密称取黄芪皂苷Ⅳ对照品适量,加甲醇溶解并定容,得198 mg/L的标准储备溶液;准确移取一定量标准储备溶液,用甲醇配制质量浓度为0.387 0、0.774 0、1.548、3.095、6.190、12.38、24.75 mg/L的系列标准工作液。
1.3 样品前处理
将药材粉碎后过四号筛(65目,国家标准R40/3系列),精密称取样品粉末2 g,提取溶剂为甲醇,将直接提取后的溶液和提取后除杂的溶液过滤后,经UPLC-MS/MS检测,外标法定量分析。
1.3.1 提取方式考察考察3种提取方式:A、将样品粉末置于索氏提取器中,用甲醇冷浸过夜,索氏提取(85 ℃) 4 h,定容至100 mL;B、将样品粉末置于索氏提取器中,用甲醇直接提取(85 ℃)5 h,定容至100 mL;C、超声提取3次,每次加50 mL甲醇,提取时间分别为1、1、0.5 h,将提取液合并浓缩后定容至100 mL。按上述提取方式制备提取液,过滤后待测。
1.3.2 净化过程考察选择Y1样品粉末按提取方式B进行提取,考察两个提取温度(85、95 ℃),每个温度做4个平行样,提取溶液均定容至100 mL,取0.5 mL过滤后待测(溶液a);剩余溶液蒸干后按《中国药典》要求,残渣加10 mL水,微热使其溶解,用水饱和的正丁醇振摇提取4次,每次40 mL,分别合并上层正丁醇液;分别将同一提取温度下的2个平行样,直接蒸干正丁醇层(溶液b);剩余2个平行样,将正丁醇层用氨试液充分洗涤2次,每次40 mL,弃去下层氨液,正丁醇层蒸干(溶液c);蒸干后(蒸干温度为95 ℃)的平行样,分别用甲醇定容至100 mL。
1.4 测定条件
色谱条件:色谱柱:Agilent HPH C18(50 mm×2.1 mm,1.9 μm);柱温:35 ℃;流动相:0.1%甲酸水溶液(A)-乙腈(B);进样量:1 μL;流速:0.35 mL/min;梯度洗脱程序:0~10 min,10%~32% B;10~15 min,32% B;15~20 min,32%~90% B;20~20.5 min,90%~10% B;20.5~22 min,10% B。
低分辨质谱条件:AJS 电喷雾离子源(ESI)正离子模式;干燥气温度:325 ℃;干燥气流速:10 L/min;鞘气温度:350 ℃;鞘气流速:11 L/min;雾化气压力:275.8 kPa(40 psi);碎片电压:120 V;碰撞能量:12 eV;检测方式:多反应监测模式(MRM);定量离子对:m/z785.6→143.2;定性离子对:m/z785.6→ 473.3。
高分辨质谱条件:正或负离子模式;扫描范围:m/z100~1 500;喷雾气压力:80 kPa;干燥气流速:7 L/min;干燥气温度:250 ℃;毛细管入口电压:4.0 kV;锥孔电压:20 V;采样大小为2 M(兆);累积时间为0.1 s;仪器进样前需用三氟乙酸钠进行校准,数据采集后根据已知成分的精确分子质量进行数据再校准,以确保质谱数据的准确性。
2 结果与讨论
2.1 不同提取方式的比较
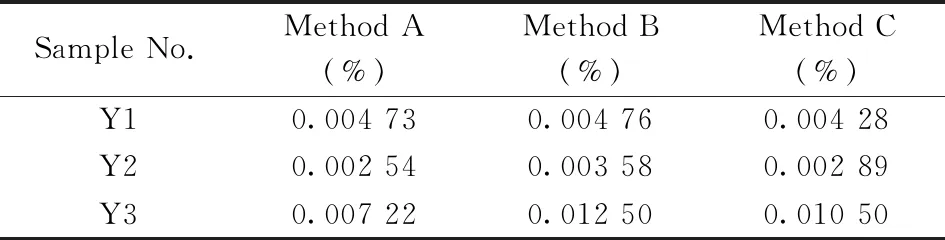
表1 不同提取方式(直接测定提取液)下 游离黄芪皂苷Ⅳ的含量Table 1 Content of free astragaloside Ⅳ under different extraction methods(direct determination of the extracts)
李焕娟等[17]对比了本文中的3种提取方式,结果表明回流提取法测得的黄芪皂苷Ⅳ含量较高。但该文献仍按照《中国药典》方法,采用了正丁醇萃取和氨试液除杂过程。本研究选取3批黄芪药材,按《中国药典》方法测定黄芪皂苷Ⅳ的含量:Y1(含0.073%)和Y3(含0.081%)样品合格,Y2(含0.019%)样品不合格。考察“1.3.1”中3种提取方式对黄芪皂苷Ⅳ提取率的影响,采用“1.4”方法测定含量。由表1可知,按《中国药典》方法提取纯化后总黄芪皂苷Ⅳ含量是直接提取后游离黄芪皂苷Ⅳ含量的5~15倍。提取方式B测定的含量最高,说明浸泡过夜不影响提取率,而提取时间直接影响提取次数,提取次数才是影响提取率的关键因素。提取方式C测得的含量较低,说明超声提取的提取率较低。相比之下,采用索氏提取法可避免繁琐操作给定量分析带来的误差。因此,本研究确定最优提取方法为方式B:采用索氏提取法,用甲醇于85 ℃提取5 h。
2.2 净化过程的影响
按“1.3.2”对Y1样品进行净化过程的影响考察,结果显示(见表2),直接提取后(溶液a)和继续用正丁醇萃取后(溶液b)黄芪皂苷Ⅳ的测定结果无显著性差异,说明蒸干提取液、正丁醇萃取和蒸干正丁醇萃取液(不加氨试液除杂)过程,均不会引起其他皂苷类成分的转化,也不会使溶液中游离黄芪皂苷Ⅳ降解。而正丁醇萃取后再用氨试液除杂(溶液c),测得的结果显著升高,与《中国药典》方法测定的结果一致,表明氨试液除杂过程会提高黄芪皂苷Ⅳ的测定结果。
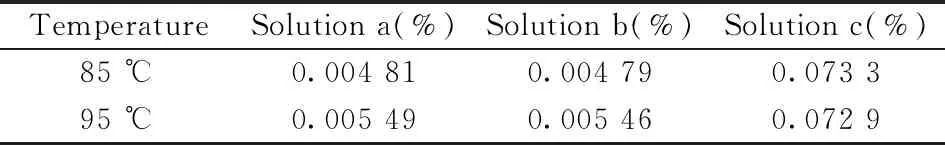
表2 Y1样品不同净化过程下黄芪皂苷Ⅳ的含量Table 2 Contents of astragaloside Ⅳ in Y1 sample under different purification processes
观察不同提取温度下索氏提取的回流情况,发现95 ℃下回流速度是85 ℃的1.5倍,因此相同时间内提取温度越高,提取次数越多。由表2可知,溶液c在85 ℃和95 ℃下提取5 h的结果无显著性差异,可推断这两个提取温度不会影响转化后总黄芪皂苷Ⅳ的含量;而溶液a和b的结果有显著性差异,说明提取温度会影响样品中游离黄芪皂苷Ⅳ的含量。因此,选择索氏提取温度为95 ℃,对游离黄芪皂苷Ⅳ的提取率更高。
2.3 FT-ICR-MS高分辨质谱分析净化过程中相关成分的差异性
将上述95 ℃下提取的溶液a、b和c分别用UPLC-FT-ICR-MS高分辨质谱进行分析,每个溶液重复测定3次,并分别采集正、负离子模式下的总离子流(TIC)图。从负离子模式下的TIC图(见图1)可见有4个峰的强度变化显著,其中与对照品的保留时间及其精确分子质量比对后确认:1号峰为毛蕊异黄酮葡萄糖苷(4.1 min,C22H22O10,理论精确分子质量为:491.118 40[M+HCOO]-/937.239 70[2M+HCOO]-);2号峰为黄芪皂苷Ⅳ(12.7 min,C41H68O14,理论精确分子质量为:829.458 01[M+HCOO]-);将精确分子质量、保留时间及其碎片离子与相关文献[8-9]进行比较,3号、4号和5号峰分别指认为黄芪皂苷Ⅱ(15.1 min,C43H70O15,理论精确分子质量为:871.468 58[M+HCOO]-)、黄芪皂苷Ⅰ(16.8 min,C45H72O16,理论精确分子质量为:913.47814[M+HCOO]-)和丙二酰黄芪皂苷Ⅰ(17.3 min,C48H74O19,理论精确分子质量为:953.474 06[M-H]-)。

图1 提取温度为95 ℃时不同净化过程的TIC图和EIC图Fig.1 Total ion chromatograms and extracted ion chromatogram of different purification processes at the extract temperature of 95 ℃ solution a:after direct extraction;solution b:after n-butanol extraction;solution c:after impurity removal by ammonia solution
比较相对峰强度后,发现黄芪皂苷Ⅱ在溶液b中的相对含量增加,而在溶液c中的相对含量降低,说明有些成分不稳定受热易水解为黄芪皂苷Ⅱ,黄芪皂苷Ⅱ在碱性条件下也会水解。丙二酰黄芪皂苷Ⅰ在正丁醇萃取后(溶液c)的含量显著降低,说明其在中性条件下不稳定。相比其他成分,黄芪皂苷Ⅰ在溶液a中的相对含量最高,在氨试液洗涤后其相对含量显著降低,同时黄芪皂苷Ⅳ含量显著升高,由此可认为黄芪皂苷Ⅰ转化为黄芪皂苷Ⅳ的贡献最大。史静超等[18]分别测定了16批蒙古黄芪中4种主要皂苷的含量,其前处理过程仅用甲醇索氏提取,结果发现黄芪皂苷Ⅰ和黄芪皂苷Ⅱ的含量均较高,其中黄芪皂苷Ⅰ的含量最高,约为黄芪皂苷Ⅳ的10~30倍,其结论与本实验结果一致。Chu等[9]研究发现,中性条件下,丙二酰黄芪皂苷Ⅰ会部分水解为黄芪皂苷Ⅰ;碱性条件下,黄芪皂苷Ⅱ、黄芪皂苷Ⅰ和丙二酰黄芪皂苷Ⅰ均会部分水解为不含酰基的黄芪皂苷Ⅳ,而丙二酰黄芪皂苷Ⅰ还会部分水解为黄芪皂苷Ⅱ。从唐冕等[19]总结的化学成分可知,黄芪药材中存在大量含有酰基的皂苷类成分,而碱处理过程会导致上述成分脱掉酰基,水解为其他成分,从而不能反映黄芪药材中原始成分的情况。因此,在研究黄芪药材中原始成分时,提取工艺不能加碱液,且在保证有效提取率的情况下,应尽量缩短提取时间;为提高黄芪药材中药效成分黄芪皂苷Ⅳ的含量,可考虑用适当的碱液对其进行炮制。
结合本研究的结果,得到其转化途径如图2所示。目前已报道[20-21]黄芪药材中存在黄芪皂苷Ⅰ、异黄芪皂苷Ⅰ和新黄芪皂苷Ⅰ这3个乙酰基在木糖上取代位置不同的异构体,而黄芪皂苷Ⅱ(乙酰基在木糖上的1位)的异构体只报道了1个为异黄芪皂苷Ⅱ(乙酰基在木糖上的2位)。对溶液c提取黄芪皂苷Ⅱ对应的离子,得到的提取离子(EIC)图(见图1)中存在分离度较好的3个峰,因此可推断黄芪药材中还存在1个乙酰基在木糖上3位的黄芪皂苷Ⅱ异构体,将其命名为新黄芪皂苷Ⅱ。
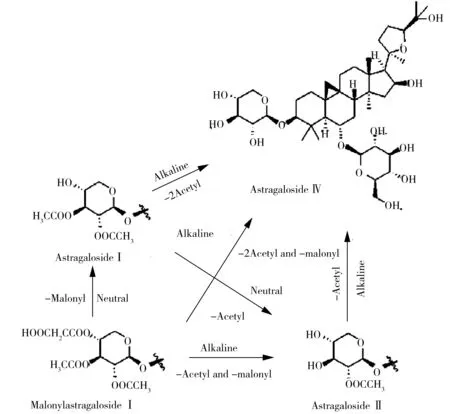
图2 中性或碱性条件下4种黄芪皂苷之间的转化途径Fig.2 The proposed transformation pathways between the four kinds of astragaloside in neutral and alkaline solutions
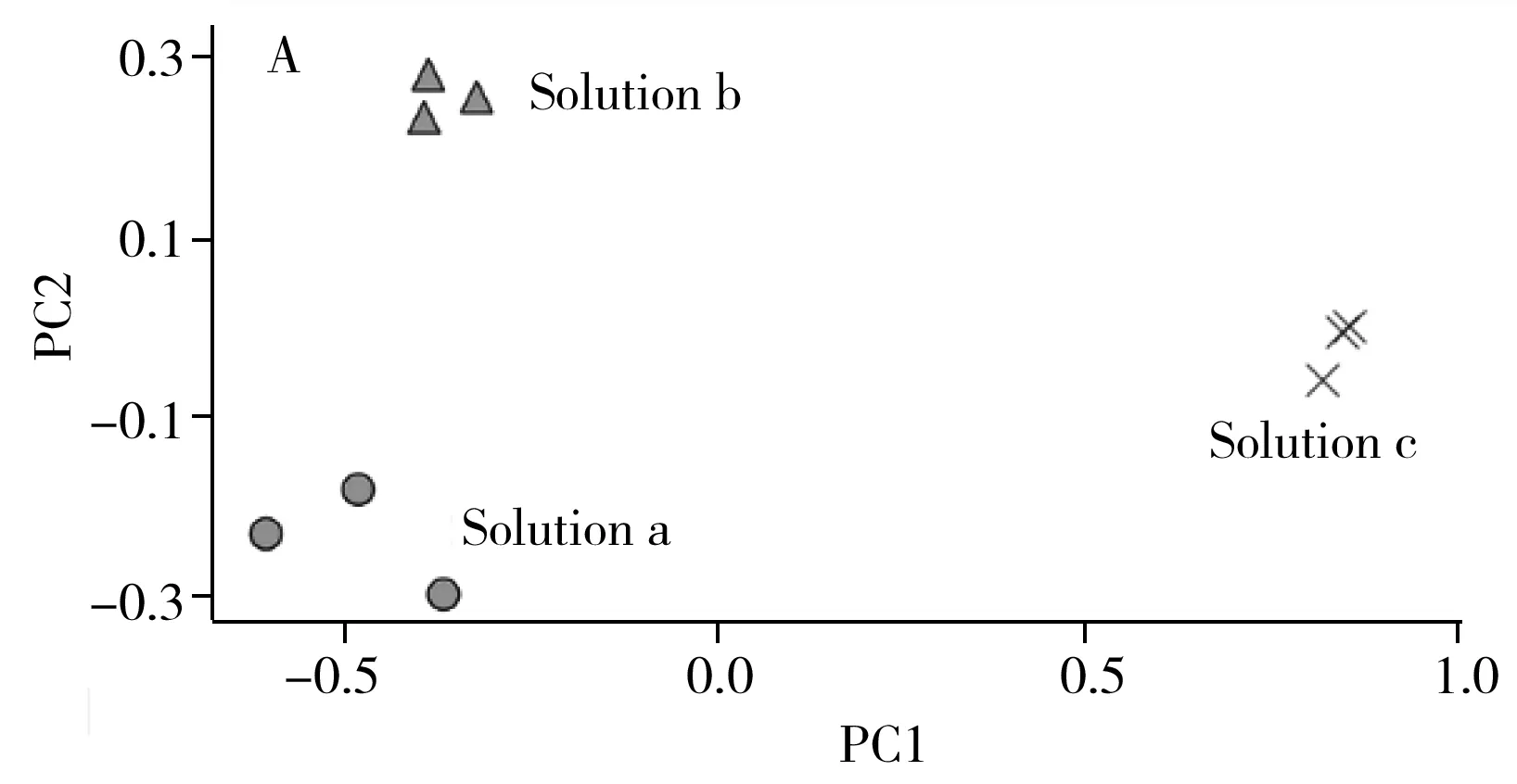
采用主成分分析(PCA)对上述UPLC-FT-ICR-MS数据进行统计学分析,在95%的置信区间下,根据PCA得分图(图3A)和荷载图(图3B)所示,溶液a、b和c的数据点被清晰分离。其中溶液a和b均落在负相关区域,而溶液c处于正相关区域,说明正丁醇萃取除去的成分相对较少,氨试液洗涤除去的成分相对较多。根据质荷比的偏离程度(图3B),得到主要影响得分结果的质荷比分别为:m/z491.12(毛蕊异黄酮葡萄糖苷)、829.46(黄芪皂苷Ⅳ)、871.47(黄芪皂苷Ⅱ)、913.48(黄芪皂苷Ⅰ),该结果进一步确认了上述4种成分为溶液a、b和c的主要差异性成分。毛蕊异黄酮葡萄糖苷是黄酮类成分,易溶于碱性溶液,因而正丁醇层的毛蕊异黄酮葡萄糖苷会被氨试液洗涤后除去。
2.4 低分辨质谱条件的优化
将质量浓度为1.0 mg/L的黄芪皂苷Ⅳ标准溶液在正、负离子模式下分别进行质谱一级全扫描,选出基峰强度较高的母离子进行二级质谱分析,最后选择在正离子模式下[16]进行条件优化。黄芪皂苷Ⅳ的[M+Na]+峰m/z807.5响应较高,但选定该母离子做二级质谱时,找不到响应较高的定性和定量离子。因此,本实验选择[M+H]+峰m/z785.6为MRM扫描的母离子,以响应较高的m/z143.2为定量离子,以m/z473.3为定性离子。此条件下,黄芪皂苷Ⅳ的低分辨质谱图及特征碎片结构如图4所示。
2.5 色谱条件的优化
本实验选择乙腈-水流动相体系,由于流动相中加入甲酸有助于[M+H]+的形成,可提高响应并改善峰形[22],因此考察了水相分别添加0.1%甲酸和10 mmol/L甲酸铵对待测物分离效果和响应强度的影响。结果表明,水相中加入0.1%甲酸后,黄芪皂苷Ⅳ的峰形相对较好,且灵敏度高,故选择乙腈-0.1%甲酸水溶液作为流动相。
考察了不同型号的3种C18色谱柱Agilent Poroshell 120 EC-C18(3.0 mm×100 mm,2.7 μm)(柱A)、Agilent Poroshell 120 SB-C18(100 mm×2.1 mm,2.7 μm)(柱B)和Agilent HPH C18(50 mm×2.1 mm,1.9 μm)(柱C)的分离效果。结果显示,在一定梯度洗脱程序下,这3种色谱柱均能得到很好的分离效果,而使用色谱柱C所需分析时间最短,因此选择Agilent HPH C18(50 mm×2.1 mm,1.9 μm)作为本方法的分离柱。
2.6 线性范围、检出限与定量下限
按照“1.4”条件对配制的系列标准工作液进行UPLC-MS/MS测定,以对照品的质量浓度(x,mg·L-1)为横坐标,所得峰面积(y)为纵坐标,绘制标准曲线。结果显示黄芪皂苷Ⅳ在0.387 0~24.75 mg/L范围内具有良好的线性关系,回归方程为y=3 633x-543,相关系数(r)为0.999 8。以信噪比S/N=3计算检出限(LOD)为1.0 mg/kg;以信噪比S/N=10计算定量下限(LOQ)为3.0 mg/kg。
2.7 回收率与相对标准偏差
取Y1样品,分别进行低、中、高3个浓度水平的加标回收实验,索氏提取前,分别准确加入198 mg/L的黄芪皂苷Ⅳ标准储备溶液0.2、1、5 mL,在95 ℃下用甲醇索氏提取5 h,提取溶液均定容至100 mL;另取6份Y1样品,采用上述提取条件进行精密度实验。结果显示,低、中、高3个加标水平下的回收率为94.5%~105%,6份平行样品的相对标准偏差(RSD)为1.4%,表明该方法具有较好的准确度和精密度,适用于黄芪药材中黄芪皂苷Ⅳ的含量测定。
3 结 论
本研究发现黄芪皂苷Ⅳ含量测定的前处理过程中氨试液除杂会导致大量黄芪皂苷Ⅳ同系物水解为黄芪皂苷Ⅳ,从而显著提高黄芪皂苷Ⅳ的含量。其中,甲醇直接提取测得的黄芪皂苷Ⅰ相对含量最高,且在碱处理后可彻底水解为具有多种药理活性的黄芪皂苷Ⅳ。按2015年版《中国药典》测定的不是游离黄芪皂苷Ⅳ,还包含其他皂苷类成分水解后的含量。此外,还发现黄芪药材中存在目前尚未报道的黄芪皂苷Ⅱ异构体,将其命名为新黄芪皂苷Ⅱ。本研究建立的黄芪药材中游离黄芪皂苷Ⅳ含量的测定方法,具有操作简单、灵敏度高、稳定可靠等特点,可准确反映黄芪药材的质量情况。
猜你喜欢
承德医学院学报(2022年2期)2022-05-23
中国测试(2022年4期)2022-05-10
海南热带海洋学院学报(2021年5期)2021-11-07
森林工程(2021年4期)2021-08-23
中成药(2018年9期)2018-10-09
中成药(2018年7期)2018-08-04
中成药(2018年3期)2018-05-07
学术交流(2018年7期)2018-02-20
移动信息(2017年3期)2017-12-28
速读·中旬(2016年9期)2017-05-09
