
X连锁charcot-Marie-Tooth病GJB1基因突变分析
2020-12-25 06:18谭玉杰
中国计划生育学杂志 2020年7期
张 婵 谭玉杰 王 芳
郑州大学附属洛阳中心医院(471000)
1 临床资料
先证者男性,30岁,20年前发现左脚行走无力,脚尖无法用力。随后3年逐渐出现双手大小鱼际萎缩,力弱,双手指关节弯曲,中指和无名指不能并拢,伴双上肢麻木、抽筋感。10年前双小腿轻度萎缩,双下肢浅感觉轻度减退,随着病程进展双下肢远端肌无力及腱反射减低,双足趾背屈不能,趾关节弯曲,行走时步态不稳,患者听力在发病期间有所减弱。精神、智商正常。体格检查:神志清楚,颅神经检查正常。颈软,心、肺听诊无异常,双上肢肌力、双下肢肌力IV级,四肢肌张力正常,腱反射减弱。辅助检查:血清肌酶谱未见明显异常。左下肢胫、腓神经及左侧胫前肌、腓肠肌肌电图显示:左侧胫腓神经损害(运动传导速度减慢,感觉传导速度正常)。全外显子检测分析:GJB1基因C.623A>G突变。该位点在人群中发生频率极低,经SIFT和polyphen软件对其进行蛋白质功能预测,结果均为有害。
追踪家系内成员,先证者母亲,55岁,10年前出现左侧手指指关节轻度弯曲,不伴功能障碍。左下肢轻度萎缩,步态稳。左足远端感觉减弱,不伴趾关节畸形。先证者哥哥,37岁,5年前出现双下肢麻木,肌肉轻度萎缩,双下肢浅感觉减退。双足趾背屈不能,趾关节弯曲,鹤腿样明显。其哥哥生育一男孩,目前无异常。姐姐,32岁,目前无异常表现。生育一男孩一女孩,目前无异常表现。家系图见图1。
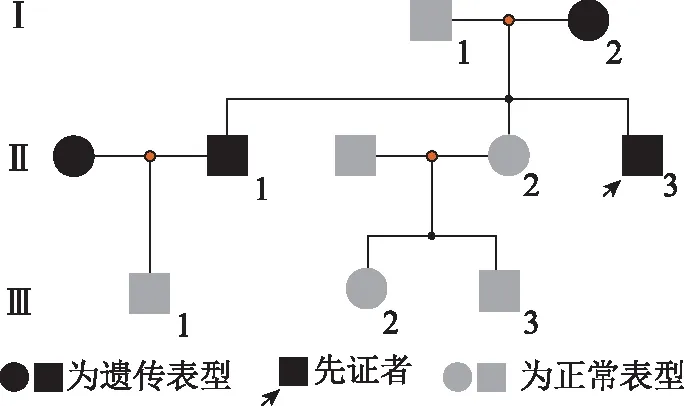
图1 X连锁charcot-Marie-Tooth家系图
2 讨论
Charcot-Marie-Tooth病(CMT)又称遗传性运动感觉神经病(HMSN)或腓骨肌萎缩症,GJB1基因突变引起的CMTXD型发病率占CMT患者的7%~11%[1]。CMT临床诊断分型主要依靠患者临床特征、电生理特征、临床病理特征等资料进行判断。由于CMT临床表型复杂,不容易被确诊。而CMTXD患者可有典型的CMT的症状,多数都是青少年发病,男性症状重于女性,下肢病变为主。CMTX位点X染色体Xq13.1,编码一种相对分子质量32000间隙连接蛋白。该蛋白在周围神经的施旺细胞和中枢神经的少突状细胞中编码[2]。该家系出现的患者男性多且症状重,遗传方式为X连锁隐形遗传。家系中女性症状较轻或没有临床症状,与X染色体莱昂化作用有关[3]。
CMT是一组高度遗传异质性疾病,笔者对先证者的全外显子测序发现该先证者存在GJB1基因的C.623A>G(p.p.Glu208Gly)致病性突变,国内尚未见有该突变报道,该突变是国内新发现的CMTX突变位点。GJB1基因全长10.3kb,包含2个外显子,其中第2外显子包含蛋白阅读框架(283个密码子),其编码的CX32蛋白是间隙连接蛋白家族的一种,有4个跨膜区、2个胞外环,1个胞内环,C端和N端均位于胞内[4],其作用是在细胞内聚合成六聚体结构八项转运至细胞膜形成半通道,再与相邻细胞膜上相同或不同半通道形成完整的间隙连接通道,容许细胞间离子、小分子、营养物质快速通过,起细胞通讯作用[5-6]。本研究中先证者2号外显子C.623A到G的错义突变导致第208位谷氨酸转变为甘氨酸。该先证者GJB1突变可能导致CX32蛋白正常功能丧失和(或)获得毒性功能,引起髓鞘功能改变和髓鞘-轴索间相互作用异常,导致周围神经病变[7-8]。CMT除周围神经病变还可出现中枢神经异常[9],脊柱畸形和感觉神经性耳聋[10]。
根据患者症状、体征及相关肌电图、全外显子测序测序结果,CMT诊断明确,询问病史发现该患者出现有近几年听力下降状况,考虑出现中枢神经系统损害症状。目前尚无逆转该病病程的治疗方法,疾病预后差。在随后此病研究中增加对该家系其他成员的检测,进一步明确该疾病的基因位点,为该家系成员的遗传咨询提供合理的指导,同时也为优生提供临床依据。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国现代医生(2022年21期)2022-08-22
临床输血与检验(2022年3期)2022-06-22
当代医药论丛(2021年24期)2022-01-20
山东医药(2021年24期)2021-09-01
诊断学(理论与实践)(2020年1期)2020-04-28
郑州大学学报(医学版)(2019年3期)2019-06-03
中南林业科技大学学报(2019年4期)2019-04-08
森林工程(2018年1期)2018-05-14
