
利用CRISPR/Cas9系统构建人HIF-1α基因敲除细胞株
2020-12-21 07:24张宝云王凭青向远彩
生物学杂志 2020年6期
杨 芳, 张宝云, 向 伟, 王凭青, 向远彩
(1. 西南医科大学 基础医学院, 泸州 646000; 2. 重庆大学 生物工程学院, 重庆 400000)
低氧诱导因子(Hypoxia inducible factor,HIF)是哺乳动物细胞内存在的一类介导低氧适应性反应的转录因子,能够激活响应低氧应激基因的表达。研究揭示,介导此过程的主要分子是HIF-1,它由敏感型的α亚基和组成型的β亚基构成异二聚体[1-2]。由于HIF-1能够参与卵泡的发育、成熟、排卵以及随后的黄体早期发育等排卵事件,因而它对哺乳动物排卵过程的正常进行具有重要作用[3-4]。除了在正常排卵过程中扮演着重要角色外,HIF-1还能促进卵巢肿瘤细胞转移及其发展[5-6],因而众多研究者[7-8]认为HIF-1可能是切断卵巢肿瘤转移的潜在靶点。HIF-1既能维持正常的排卵过程又能参与如卵巢癌、子宫内膜癌等肿瘤的发展,这种看似矛盾的作用机制恰恰提示其在卵巢中的重要性。因此,了解HIF-1如何在不同细胞的不同时期对卵巢进行精准调控具有重要的研究价值。为了探明HIF-1在卵巢中的复杂调控机制,亟需获得高效快速的方法在不同细胞中构建HIF-1功能缺失的稳定细胞株。
基因编辑技术是一项以基因靶向修饰为基础的可对生物基因组进行改造的新技术。随着近几年的发展,基因编辑技术有了巨大的飞跃,目前CRISPR/Cas9系统因其便捷、高效的特性已逐渐取代传统的TALEN和ZFN技术,并广泛运用于人类细胞、斑马鱼、小鼠以及细菌的基因组等精确修饰[9-13]。因此,本实验采取CRISPR/Cas9技术靶向编辑HIF-1敏感性α亚基(HIF-1α)的策略来达到失活HIF-1的目的:首先将编辑质粒转入KGN细胞,用嘌呤霉素筛选出特异性敲除细胞株并对其鉴定;再用氯化钴模拟低氧环境处理正常和敲除细胞株,检测HIF-1下游基因的表达,以验证HIF-1α缺失后对HIF-1功能的影响。
1 材料与方法
1.1 材料
KGN细胞由本实验室保存(成都中科院赠予);pCAG-T7-cas9+gRNA-pgk-Pro-T2A-GFP载体(CRISPR/Cas9敲除质粒,简写Cas9-gRNA)、引物、Oligos均购自北京唯尚立德公司;基因组试剂盒(康为世纪生物科技有限公司);嘌呤霉素(sigma公司);T7核酸内切酶I(T7E1,TAKARA公司);HIF-1α抗体(NOVUS biological);β-actin抗体(中山金桥);PPARγ(Proteintech公司)和ELISA试剂盒(百蕊生物公司)。
1.2 sgRNA设计和Cas 9-gRNA- HIF-1α表达质粒构建
在该基因的外显子序列中寻找PAM序列(NGG),选取该序列上游紧邻的20 bp碱基序列作为sgRNA。将选好的sgRNA序列在NCBI系统中进行BLAST比对,排除单核苷酸多态性的序列,根据CRISPR/Cas9敲除质粒设计说明,在sgRNA的末端加上黏性末端AAACACCG(正义链5′端)和AAACACCG(反义链5′端)。所设计的sgRNA序列为:HIF-1asg-sense:5′-AAACACCGAAGTGTACCCTAACTAGCCG-3′;HIF-1asg-anti:5′-CTCTAAAACCGGCTAGTTAGGGTACACTT-3′。将设计好的Oligo经退火形成二聚体后,与质粒骨架连接并进行转化。挑3~5个白色菌落摇菌,提取质粒并测序验证,测序引物序列为:5′-TGAGCGTCGATTTTTGTGATGCTCGTCAG-3′。
1.3 细胞转染
将一定数量KGN细胞种于6孔板中,待细胞密度达到70%~80%时进行转染。转染时,将2 μg Cas9-gRNA-HIF-1α表达质粒与125 μL无血清转染培养基混合,同时,将5 μL转染试剂lipofectamin 3000与125 μL无血清转染培养基混合。5 min后,将两者等比例混匀,并在室温放置20 min,最后将此混合物加入到6孔板中,并用无血清转染培养基补齐至1 mL,轻晃混匀,并于37 ℃培养8 h后更换为正常培养基,24 h后可进行荧光检测和药物筛选。
1.4 细胞分选
由于Cas9-gRNA-HIF-1α表达质粒的同时表达抗性标签(Puromycin)和荧光蛋白(GFP),故细胞分选时选择药物筛选,具体如下:待转染24 h后,在荧光显微镜下观察以确定转染效率,随即加入嘌呤霉素(2.5 μg/μL)进行药筛,待对照组细胞完全死亡时,加入正常培养基进行恢复培养。待细胞恢复一段时间后,用胰酶消化,并采用有限稀释法将单个细胞转移到96孔板中进行培养,按正常情况更换培养基。待筛选出的单个细胞长成细胞团后逐次扩大培养至24孔板。同时,收集一部分细胞提取基因组DNA用于PCR进行目的片段扩增(引物F:5′-GCTGGAGTGAGTAAAAGGGGA-3′,R:5′-CAAAGCCAACTGATTAAGCCTGG-3′)。
1.5 T7E1酶切实验及单克隆鉴定
以基因组DNA为模板,根据gRNA两侧碱基特点设计包含该靶点的克隆引物,通过PCR获得克隆产物。将敲除组PCR产物与野生型PCR产物等量混合,并退火杂交:若产生非配对的DNA片段,那么,这些片段能被T7E1剪切;若没有发生突变,将产生配对的片段,该酶无法进行剪切。退火后,在各反应体系中分别加入0.5 μL T7E1,37 ℃反应30 min后,用2%的琼脂糖凝胶进行电泳检测,分析酶切结果,并对有剪切条带产生的单克隆PCR产物进行测序鉴定。
1.6 Western Blot检测
将一定数量的正常细胞和敲除细胞种于6孔板中,待细胞生长至80%左右时,用氯化钴处理12 h,弃培养基并用PBS清洗,随即加入100 μL裂解液(蛋白抑制剂PMSF∶RIPA裂解液=1∶100),用细胞刮反复刮擦,使细胞完全脱落后转移至无菌的离心管中,冰上孵育30 min后于4 ℃离心15 min (12 000 r/min),取上清液。加入5×上样缓冲液,100 ℃水浴10 min。用10%的SDS-PAGE凝胶进行电泳,并经转膜、封闭1 h、孵育一抗过夜、TBST漂洗、孵育二抗、TBST再漂洗及ECL显色等步骤检测目的蛋白的变化。
1.7 排卵相关基因的检测
将细胞种于6孔板,待细胞密度达到80%左右时,用氯化钴处理正常和HIF-1α特异性敲除细胞12 h后,分别提取RNA和蛋白并进行检测。RNA的检测:取出细胞,吸除培养液,PBS清洗2次,加1 mL RL裂解液,转移至离心管中,每1 mL RL加0.2 mL氯仿,离心,试剂盒提取RNA,反转录获得cDNA。按照检测基因的数目配制合适的体系,进行定量检测,分析实验结果(相关基因及引物信息详见表1)。蛋白的检测:PPARγ检测方法同1.6,Edn2则用ELISA试剂盒进行检测。
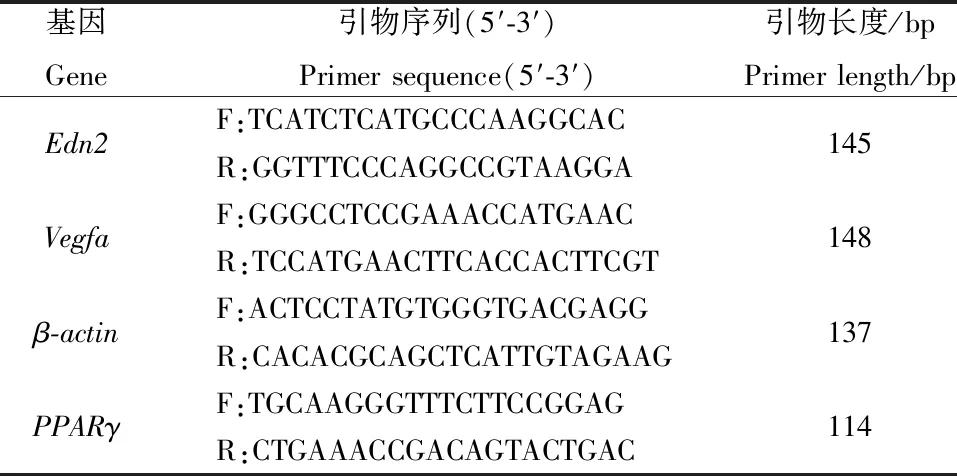
表1 排卵相关基因引物
2 结果与分析
2.1 sgRNA的选取及质粒的构建
在NCBI中查找HIF-1α(Gene ID: 3091),发现该基因位于14号染色体q23.2区域,编码区域长52 746 bp。其中,包括16个外显子和15个内含子,编码826个氨基酸,蛋白质大小约为120 ku。已有报道[14-16]显示,HIF-1α通过PAS结构域(Per-ARNT-Sim domain,93-339 aa)与HIF-1β结合以二聚体的形式发挥作用。为此,在HIF-1α的PAS区域内寻找靶点以破坏HIF-1α的功能。依据设计原则,最终在Exon 5区域内发现适合的靶序列AAGTGTACCCTAACTAGCCG(对应195-201 aa),该靶点位于PASa(106-162 aa)与PASb(249-300 aa)之间[图1(a)和(b)],理论上具有可行性。将设计好的Oligo(HIF-1αsgRNA)退火以形成Oligo二聚体,并与Cas9-gRNA载体连接后(即Cas9-gRNA-HIF-1α),经转化、涂板、测序验证[图1(c)]。
2.2 HIF-1α基因敲除细胞株构建
将Cas9-gRNA-HIF-1α质粒转染到KGN细胞中,用荧光显微镜观察,发现该质粒在KGN细胞中的转染效率约为20%[图2(a)]。为检测该质粒是否有编辑作用,提取转染后细胞的DNA,经PCR克隆后测序,发现在靶点序列附近出现套峰[图2(b)],而且在另一细胞系A2780中也得到类似的结果(数据未展示),这说明该质粒具有编辑HIF-1α的能力。为进一步快速获得单克隆细胞,用嘌呤霉素进行筛选。其间共筛选了21个单克隆细胞,随即通过PCR扩增编辑区域片段,并用T7E1酶切法进行酶切。若编辑细胞与野生型细胞扩增片段配对的产物能在短时间内被酶切处理后产生3条不同大小的DNA片段(从上到下依次为全长、酶切大片段、酶切小片段),则说明潜在的突变型已被成功编辑[图2(c)第3泳道];若对目标样PCR产物单独加酶处理也出现3条带,则说明该样品内存在编辑后突变的亚型,所提取的细胞来源并非来自一个单克隆或挑选的单克隆为杂合子[图2(c)第4泳道],还需要继续挑选单克隆。实验中,经T7酶切鉴定后,最终获得5株酶切较微弱的细胞株(H1、H5、H10、H11和H21)和1株有纯合子现象的细胞株(H20)。为进一步明确这些细胞中编辑HIF-1α的具体位置,分别将它们的PCR克隆产物连接于T载体后,选取10个菌落测序。测序结果显示:这6株细胞的HIF-1α均发生了编辑。其中,H1插入了1个碱基、H5缺失3个碱基且1个碱基被替换、H10缺失5个碱基、H11有6个被替换碱基、H20缺失4个碱基、H21缺失3个碱基[图2(d)]。进一步分析来源于不同细胞株的10个单克隆发现,仅H20为纯合子(表2)。
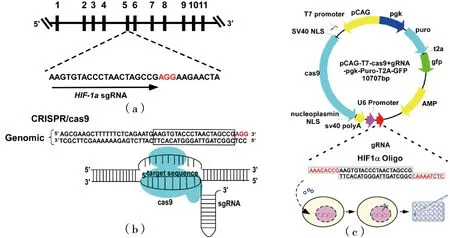
图1 sgRNA选取与质粒的构建
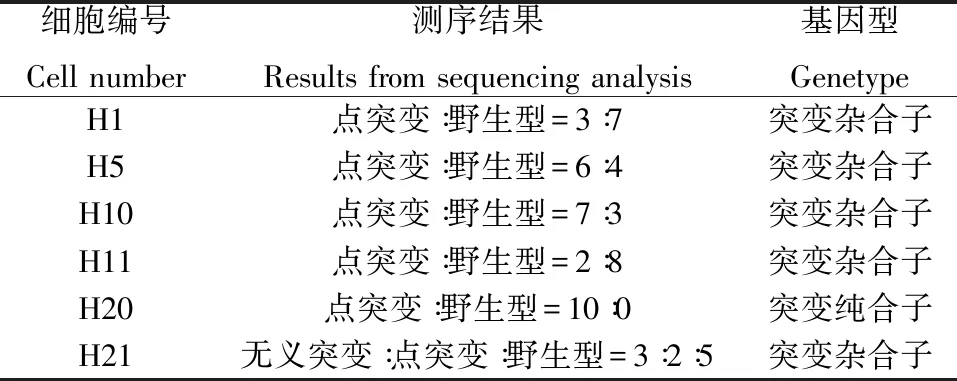
细胞编号Cell number测序结果Results from sequencing analysis基因型GenetypeH1点突变∶野生型=3∶7突变杂合子H5点突变∶野生型=6∶4突变杂合子H10点突变∶野生型=7∶3突变杂合子H11点突变∶野生型=2∶8突变杂合子H20点突变∶野生型=10∶0突变纯合子H21无义突变∶点突变∶野生型=3∶2∶5突变杂合子
2.3 Western Blot 检测HIF-1α蛋白表达
为进一步验证H20细胞中HIF-1α蛋白的表达情况,用氯化钴处理野生型(HIF-1α+/+)和敲除细胞(HIF-1α-/-)。免疫印迹结果(图3)显示,氯化钴可明显诱导野生型细胞中的HIF-1α。然而,与正常细胞相比,H20细胞中HIF-1α几乎不表达,这说明H20细胞是HIF-1α缺失的稳定细胞株。
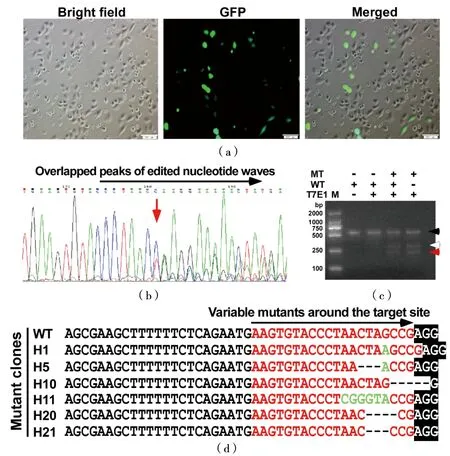
(a)荧光检测Cas9-gRNA-HIF-1α的转染效率;(b)基因编辑后,PCR扩增产物测序峰图;(c)将敲除组靶点附近的PCR产物与野生型PCR产物等量混合,并退火杂交,用T7E1酶切鉴定获得的单克隆细胞(黑色箭头所指条带表示扩增序列完全配对的野生型条带,红色和白色箭头表示T7E1酶从未完全互补配对处酶切后产生的两个片段);(d)测序后获得6组有编辑效果单克隆的序列信息
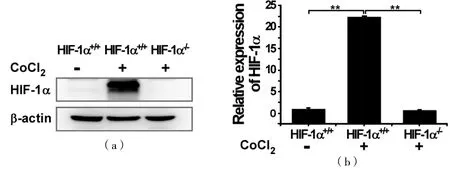
(a)用氯化钴(CoCl2)处理野生型(HIF-1α+/+)和H20细胞(HIF-1α-/-)12 h后,HIF-1α蛋白表达的免疫印迹;(b)相对定量统计。**,P<0.01
2.4 HIF-1α基因定点突变细胞株中相关基因的表达
为验证HIF-1α缺失后对HIF-1功能的影响,用氯化钴处理野生型细胞和稳定敲除细胞12 h后,检测HIF-1的下游基因Vegfa、Edn2和PPARγ的表达。实时荧光定量PCR结果显示[图4(a)~(c)]:与对照组相比,氯化钴处理后,这3个基因在野生型细胞中的表达量均显著性升高,然而,在相同情况下,敲除细胞中Vegfa、Edn2与PPARγ的表达量明显下降(其中Edn2和PPARγ最为显著)。进一步在蛋白水平上检测发现:当氯化钴处理HIF-1α+/+细胞时,Edn2和PPARγ的蛋白表达量明显增加;而在氯化钴处理HIF-1α-/-细胞时,与HIF-1α+/+细胞相比,两者的表达量显著降低,几乎与未处理的对照组一致[图4(d)和(e)]。
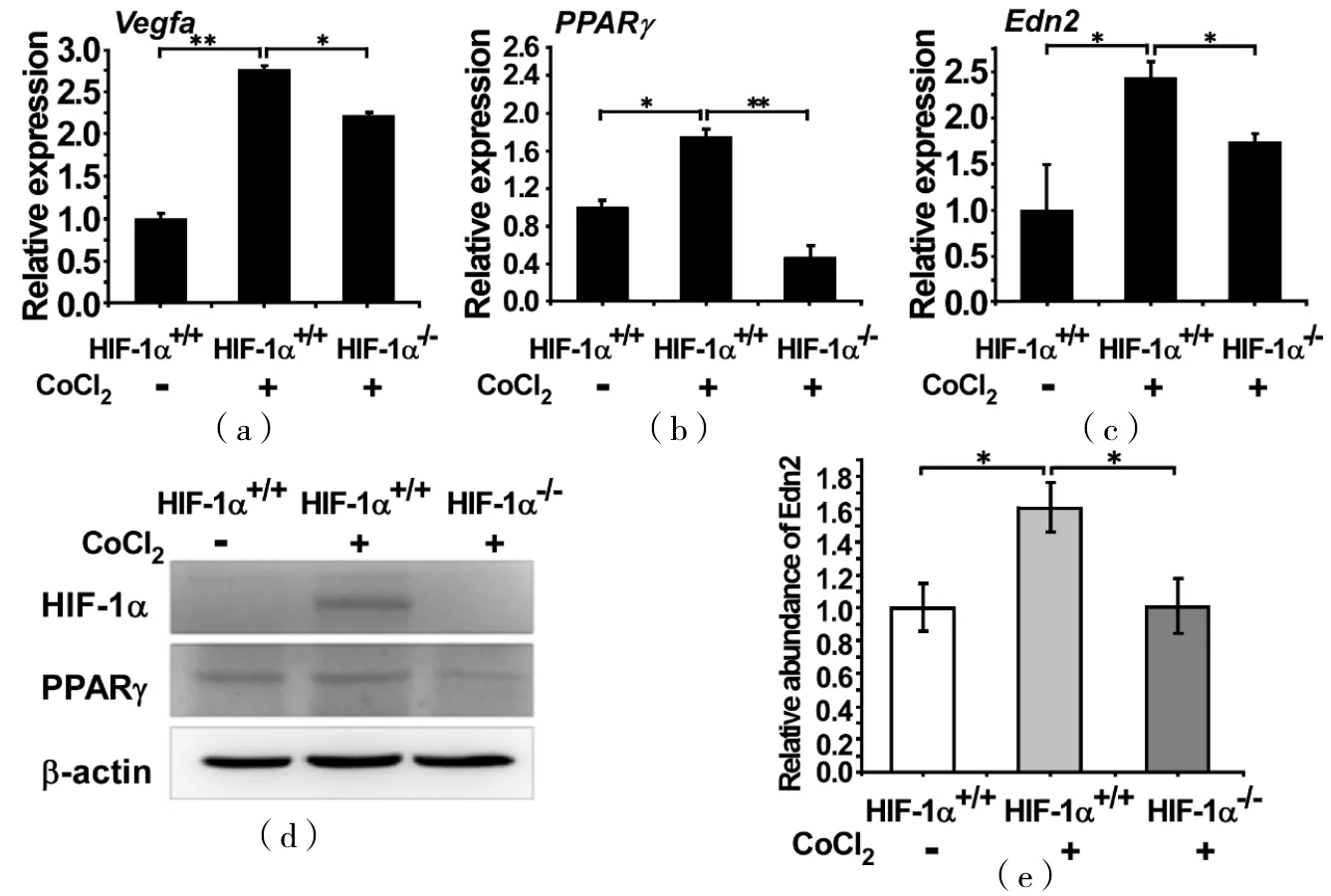
(a)~(c)收集样本后提取RNA,用定量PCR检测Vegf、PPARγ和Edn2的mRNA变化;(d)收集蛋白样品,检测HIF-1α和PPARγ蛋白表达;(e)同样的蛋白样品通过ELISA检测Edn2的蛋白表达情况。*,P<0.05;**,P<0.01
3 讨论与结论
本文主要利用CRISPR/Cas9技术靶向编辑KGN细胞中HIF-1α基因的PAS区域,通过基因编辑破坏HIF-1α的结构域,从而使HIF-1功能失活。经T7核酸内切酶I酶切、测序比对、检测目的基因及蛋白表达,最终挑选出一株纯合子细胞株(H20)。
所用CRISPR/Cas9系统的组成较为简单,只需要单个Cas9蛋白、PAM位点(通常为NGG)和gRNA进行识别编辑[10, 17]。用该技术进行基因编辑时,通过gRNA识别靶位点,并由Cas9蛋白结合至靶点进行切割,产生DNA双链缺口,进而启动两种自身修复机制——非同源末端连接和同源重组修复。其中,非同源末端连接在DNA修复中发挥主要作用,由于在修复连接时易发生碱基插入或缺失突变,因而,常用于单位点基因敲除或大片段的删除,这一点在单克隆测序结果中也能得到体现。鉴于KGN细胞的转染效率较低,用嘌呤霉素筛选已编辑的细胞,并通过有限稀释法挑选出单克隆细胞。该方法与病毒转染、流式细胞仪筛选相比,相对简单,易于操作。为最大程度地对HIF-1α进行编辑,将CRISPR/Cas9系统编辑靶点选定于靠近氨基端的PAS区域,试图通过编辑使该区域发生移码突变达到失活HIF-1α的目的,获得了HIF-1α因缺失4 bp而发生移码突变的纯合细胞株(H20)。用免疫印迹检测时发现,该细胞中的HIF-1α几乎完全缺失,与此同时,其参与调控的下游基因PPARγ和Edn2也显著性降低,这说明选择该靶点对HIF-1α敲除具有较强的可行性。需要说明的是,在检测Edn2蛋白水平的过程中,通过免疫印迹法并未检测出Edn2,这可能与其在细胞中的本底水平较低有关,抑或是Edn2蛋白质本身不稳定,迅速降解导致其蛋白水平低而无法检测,具体原因有待进一步验证。
值得一提的是,CRISPR/Cas9系统的出现给基因研究带来了光明的前景,目前,该系统已开发出病毒性、非病毒性和物理介导等多种导入方式以适用于不同的研究[18]。尽管如此,其在基因编辑过程中易出现插入突变、致免疫性和脱靶等的特性仍然极大地限制了CRISPR/Cas9系统的运用[13]。有研究人员[19-20]新开发了一种高精确度的基因编辑技术——单碱基编辑技术,无需依赖DNA模板便可有效实现数种单碱基的自由转换和多碱基的精准插入与删除,这或许为解决目前CRISPR基因编辑技术的瓶颈打开了一扇窗,值得进一步关注。
本研究在HIF-1α的PAS区域设计特异性靶点,挑选出HIF-1α特异性敲除细胞株,为在其他细胞中通过CRISPR/Cas9系统特异性敲除HIF-1α提供方法借鉴,同时也为继续探究HIF-1在由正常排卵事件演变成卵巢肿瘤过程中的作用机制奠定基础。
猜你喜欢
中国现代医药杂志(2020年10期)2020-12-14
教学考试(高考生物)(2020年6期)2020-11-23
食品与生物技术学报(2020年8期)2020-01-06
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
中成药(2018年3期)2018-05-07
现代检验医学杂志(2016年3期)2016-11-15
医学研究杂志(2015年11期)2015-06-10
医学研究杂志(2015年3期)2015-06-10
特产研究(2015年1期)2015-04-12
