
儿童肝豆状核变性一家系的基因诊断及分析
2020-12-04 08:23:08杨文明
中国实用神经疾病杂志 2020年21期
杨 悦 杨文明 李 祥
1)安徽中医药大学研究生院,安徽 合肥 230038 2)安徽中医药大学第一附属医院,安徽 合肥 230031 3)新安医学教育部重点实验室,安徽 合肥 230038
肝豆状核变性是由于铜代谢障碍导致肝脏、脑部、肾脏、角膜等组织脏器损害的神经系统遗传性疾病,因Wilson对本病进行初次报道,故本病又名Wilson 病(Wilson’s disease,WD)。流行病学资料显示[1-3],WD在世界范围的发病率<1/10万,而我国发病率高达6/10万,其致病基因携带率约为1/90。由于铜沉积的组织器官不同,因此临床上WD患者临床表现多种多样。但是根据其损害部位特征,可主要分为肝型、脑型、混合型及其他类型四种。肝型患者主要临床症状包括无症状性转氨酶升高、肝肿大、脾肿大、肝炎、脂肪肝、肝硬化和急性肝衰竭等[4]。脑型患者主要表现包括运动障碍、认知障碍、精神障碍等[5]。除肝脏和神经系统症状外,过量的铜的还会沉积于肾脏、骨骼关节、血液、皮肤等其他组织器官中,引起相应的组织器官损伤,可表现为蛋白尿、血尿、全身水肿、骨折、关节疼痛、急性溶血性贫血、皮肤色素沉着等[6]。当铜异常堆积在角膜后弹力层Descemet膜上堆积形成棕褐色、黄绿色的色素环(Kayser-Fleischer rings,K-F环),K-F环的存在高度提示本病的可能[7]。
WD是在线人类孟德尔遗传数据库(OMIM)中少数通过积极治疗而具有良好长期预后的神经系统罕见遗传病,早期诊断和规范化治疗具有重要意义。但是由于儿童WD临床症状常不典型,常规检测方法对非常年幼的儿童可能不敏感,临床确诊率低、误诊率较高。因此欧洲儿童WD指南推荐使用基于症状的诊断评分系统,全面综合评估以尽早明确诊断[8-9]。该指南同时指出,对于有症状的儿童应在2~3岁时通过家庭筛查确定诊断后立即开始治疗,以延缓疾病的进展,其具体治疗方案应针对器官受累的类型和严重程度进行个性化调整。本研究通过对一个儿童WD可疑家系进行致病基因鉴定,及时明确诊断并进行早期治疗,避免了误诊和误治。
1 资料与方法
1.1临床资料研究对象为2019-07就诊于安徽省中医院神经内科的一个WD 可疑家系。其中先证者为双胞胎姐姐,4岁。6个月前患儿体检时发现转氨酶升高,但外院查病毒性肝炎、自身抗体检测正常,病程中无任何不适症状。曾不规律服用保肝类药物,肝功能未恢复正常。外院疑诊WD,现就诊于我科为求明确诊断。家族史中父母无血缘关系、非近亲结婚,先证者双胞胎妹妹有相同病史,其家系遗传系谱图见图1。患儿生长发育如常,入院体检:肝、脾均未触及,心肺及神经系统无异常。辅助检查:(1)肝功能:ALT 63 U/L,AST 58 U/L,胆红素及白蛋白正常;肝脏超声内部回声增强增粗、分布不均匀;(2)铜代谢指标:铜蓝蛋白(CER);0.15 g/L;铜氧化酶:0.24 活力单位/L;血清铜:1.75 μmol/L;24 h 尿铜:284.73 μg;(3)裂隙灯下未见角膜色素环。先证者双胞胎妹妹检查结果基本相似均提示肝功能异常和铜代谢障碍。先证者父亲:肝功能正常,CER 0.25 g/L。先证者母亲:肝功能正常,CER 0.30 g/L;先证者弟弟:ALT 148 U/L,AST 101U/L,CER 0.13 g/L。
根据Ferenci评分[10],先证者CER为0.15 g/L,介于0.1~0.2 g/L,记1分;24 h 尿铜值>200 μg/24 h,记2分。目前总评分为3分,WD诊断可疑。除上述检查外,WD诊疗指南还推荐青霉胺负荷实验、肝组织活检、肝铜测定以及基因检测作为其诊断方法。但由于青霉胺负荷实验灵敏度低,肝组织活检及肝铜的测定均为有创检查,而基因诊断具有准确、快速、安全等优势,故本家系采用基因检测技术以协助该家系明确诊断。本院伦理委员已批准本研究,且所有家系中所有成员均已知情同意。
1.2方法
1.2.1 样本采集及 DNA 提取:用含有EDTA的真空采血管完成5人外周血采集后,先用北京天根生化科技有限公司的全血基因组DNA提取试剂盒进行外周血DNA提取,并用德国Eppendorf的分光光度计进行DNA浓度检测,最后将所有DNA样本保存在-80 ℃冰箱。
1.2.2 基因测序:将ATP7B基因外显子及其外显子内含子连接区引物设计完成后,交由南京擎科生物有限公司合成(表1)。然后采用PCR技术先扩增先证者ATP7B外显子序列,经琼脂糖凝胶电泳验证正确后,将PCR产物送至南京擎科生物有限公司用ABI PRISM 3100测序仪测序。用Chromas软件将测序结果和UCSC 基因信息数据库中ATP7B标准基因组序列(NM_000053)进行分析比对,发现其突变位点,并在家系其他成员中进行验证(图2)。

表1 ATP7B两突变位点外显子PCR引物序列Table 1 ATP7B two mutation sites exon PCRprimer sequences
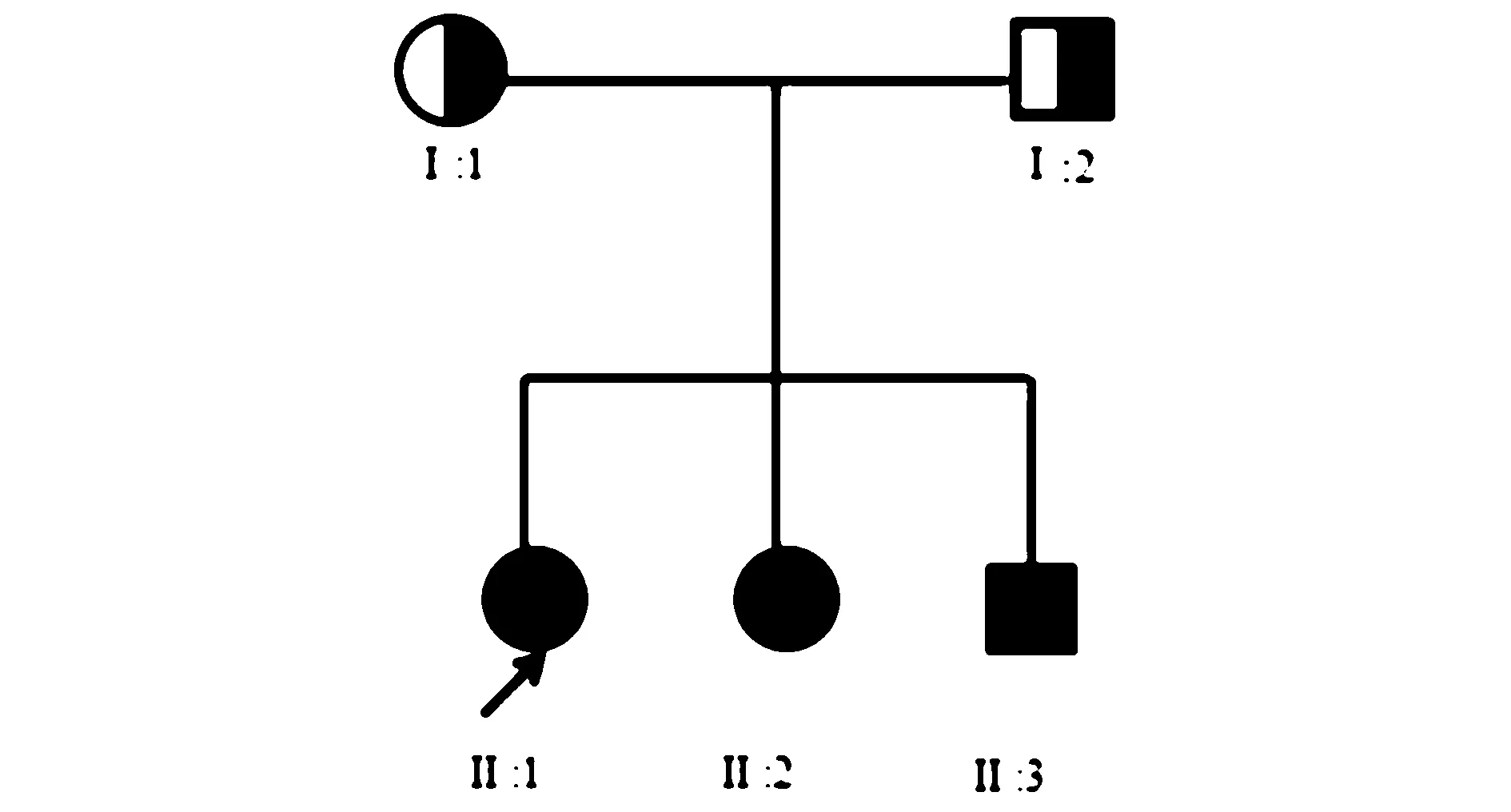
为携带致病基因女性;为携带致病基因男性;●为女性患者;■为男性患者;箭头指向先证者图1 遗传系谱图Figure 1 Genetic pedigree
2 结果
先证者的ATP7B基因存在c.2304dupC(p.Met769HisfsX26)和c.2975C>T(p.Pro992Leu)突变(图2C)。通过检索HGMD数据库、dbSNP等多个数据库证实,两突变为WD已知致病位点。c.2304dupC导致ATP7B的第769位蛋氨酸变成组氨酸并出现移码产生终止密码子使翻译提前终止。c.2975C>T导致ATP7B蛋白的第992位脯氨酸变成亮氨酸。先证者的母亲携带c.2975C>T错义突变,表型正常(图2A)。先证者父亲携带c.2304dupC移码突变,表型正常(图2B)。先证者双胞胎妹妹及弟弟ATP7B基因存在和先证者同样突变,均为WD患者(图2D、E)。

注:A-E为正向测序结果;箭头指向突变位点图2 家系测序结果图Figure 2 Sequencing results of family
3 讨论
WD是一种以肝脏和基底节损害为主的神经系统遗传病,ATP7B为本病目前已知的唯一致病基因。ATP7B是主要编码P型ATP7B的转运酶,主要分为铜离子结合区、P型ATP 酶的功能区、跨膜区3个功能区,其中跨膜区(Tm)又称跨膜转导区或疏水区,包括Tm1~8,是铜离子进出细胞膜的重要通道[11]。编码跨膜区的8号和13号外显子是中国患者突变热点[12]。本文先证者8 号外显子存在c.2304dupC移码突变,导致第769位蛋氨酸变成组氨酸并出现移码产生终止密码子使翻译提前终止,引起TM4功能区破坏。蛋氨酸属于脂肪族氨基酸,是非极性的疏水氨基酸;组氨酸属于杂环氨基酸,极性带正电荷的氨基酸。Tm4是的第4个铜离子转运的跨膜通道,此突变会引起ATP7B亚细胞定位异常,铜离子转运障碍。13号外显子存在c.2975C>T错义突变,导致第992位脯氨酸变成亮氨酸,引起Ch/Tm6功能区破坏。脯氨酸和亮氨酸均属于脂肪族氨基,是非极性的疏水氨基酸。Ch/Tm6是的第6个铜离子转运的跨膜通道,此突变会引起磷酸化异常,铜离子的正常转运受到影响[13-14]。
根据最新HGMD人类基因突变数据库记载,目前已有近800个突变已经被报道,其中以错义和无义突变最常见。ATP7B的突变热点具有地域差异,c.2333G>T(Arg778Leu) 在亚洲国家,尤其是中国WD患者中等位频率最高,而c.3207C>A(H1069Q)是目前欧洲最常见的WD突变位点[15-16]。中国WD患儿中约86.0%首发症状为无症状性转氨酶,并发现携带R778L的急性肝衰竭的发生率高于无R778L基因突变[17]。目前针对WD基因型与表型之间尚无明确关系,考虑可能与表观遗传等因素可能有关[18]。本病例中姐弟3人ATP7B基因存在c.2304dupC移码突变和c.2975C>T错义突变。有研究者报道了一名5岁的肝型WD中国女童,携带与本文先证者相同的突变位点且临床表现相似[19]。对其弟弟进一步完善相关检查显示,铜氧化酶:0.028活力单位/L;血清铜:1.10 μmol/L;24 h 尿铜:245.63 μg;裂隙灯下未见角膜 色素环。结合基因检测结果,姐弟3人最终总评分均为4分,均被明确诊断为WD患者。嘱低铜高蛋白饮食,并予以口服锌剂及排铜保肝类中药等中西医联合治疗,6个月后复查肝功能正常。
据报道儿童WD临床表现复杂多变,误诊率可高达90.5%,WD患者发病年龄2~46岁,以神经系统损害起病者发病年龄(21.5±8.2)岁,以肝脏损害起病者发病年龄(13.7±8.6)岁[20]。本病例先证者起病年龄为4岁,其弟弟起病年龄为2岁,首发症状均为无症状性的肝酶升高,临床表型均为肝型,K-F环均(-)。目前已知的最小的以无症状肝酶升高为首发症状的肝型WD患儿是1名来自日本的8个月的男童,通过基因检测发现其存在c.2621C>T(p.A874V)和c.3809A>G(p.N1270S)突变[21]。目前因为肝衰竭而导致儿童WD患者死亡病例报告越来越多,大量临床研究表明,肝硬化增加了本病死亡的风险,而早期诊断可能会增加WD患者生存时间,减少肝脏移植的需要[22]。
因此,对于肝脏受损为主的WD患儿应进行长程管理,治疗的总体目标是通过平衡铜的摄入和排泄来建立正常的铜稳态,实现净负铜平衡,延缓病情发生发展,降低死亡风险。
由于铜在不同器官沉积积累,因此WD患者临床表现各异,除了常见的肝型和脑型症状以外,现在WD相关的肾损害、骨关节病变、溶血性贫血等病例报告也层出不穷。晚期的WD患儿由于铜代谢明显紊乱而具有经典的生化特征,因此WD确诊率高而误诊率低。然而,鉴于早期患儿常规检测方法可能不敏感,并且可能无K-F环,在患有轻度肝病的无症状患儿中建立WD的诊断通常是具有挑战性的。一些中心正在使用其他检测方法以期来提高诊断水平,例如放射性标记的铜结合到在WD中受损的铜蓝蛋白中的测量、血清可交换铜,相对可交换铜[23-25],但是仍需要进一步的大样本研究来评估其在儿童WD中的诊断价值,另外也有许多专家认为基因测序技术将是克服诊断困难的好方法[26]。2018年欧洲肝病协会WD指南[27]指出,当CER 低于 0.2 g/L 时就要考虑 WD可能,当低于 0.1 g/L,应高度怀疑WD。对于不能明确的WD患者优先推荐行ATP7B基因检测,并强烈建议对其进行一级亲属行肝功能、铜代谢指标及基因检测。作为少数能够通过治疗得到有效控制的神经系统遗传疾病之一,希望通过这篇文章能够提示大家在临床中应关注早期临床症状、生物标志物,必要时使用基因测序技术,提高诊断意识,减少延误,为早期临床干预提供依据,尽早进行长程管理,从而有更好的预后。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29 01:57:52
新民周刊(2022年27期)2022-08-01 07:04:49
临床输血与检验(2022年3期)2022-06-22 02:52:50
传染病信息(2021年6期)2021-02-12 01:52:58
中国生殖健康(2020年4期)2021-01-18 02:58:10
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
中国生殖健康(2018年4期)2018-11-06 07:12:16
重庆医学(2015年12期)2015-03-05 05:52:54
生物医学工程学进展(2015年1期)2015-02-28 14:53:42
