
伴脑白质病变的强直性肌营养不良1例报告
2020-12-04 10:07:22苏春贺白宏英顾志强
中国实用神经疾病杂志 2020年21期
李 楠 张 续 苏春贺 白宏英 顾志强
郑州大学第二附属医院,河南 郑州 450003
1 临床资料
患者男,37岁。因“双下肢无力,行走不稳4 a”入院。4 a前无明显诱因渐出现肢体无力,双下肢较著,主要表现为长时间站立,爬楼梯时费力,体位改变时(下蹲站立、卧位坐起时)明显伴行走不稳,直立时出现身体前倾,向右侧偏斜,行走时步幅緩慢,向两侧摇摆,症状持续存在。自幼体质一般,学习成绩稍差,初中毕业,右眼6 a前曾行白内障手术,左眼5 a前行白内障手术。体格检查:声音低沉,秃顶,面容瘦长,颧骨隆起,呈“斧状脸”,四肢肌张力正常,双上肢肌力5级,双下肢肌力4级,双侧足背伸4级。蒙特利尔认知评估量表(Montreal cognitive assessment,MoCA)评分17分(满分30分),≤19分为异常;简易精神状态评价量表(mini-mentail state examination,MMSE)评分20分(满分30分),≤24分为异常。起立试验阳性,双侧跟膝胫试验欠稳准,蹒跚步态,走“一”字不稳。双侧桡骨膜反射减弱,双侧肱二、三头肌反射减弱,跟-膝胫反射减弱。余未见阳性体征。实验室检查:总胆固醇5.79 mmol/L,甘油三酯3.41 mmol/L,同型半胱氨酸29.1 μmol/L,HLA-B27阴性。辅助检查:心电图:(1)心电轴右偏;(2)前间壁T波改变。腰椎X线:腰椎骨质增生。彩超:右侧锁骨下动脉内中膜增厚,左侧甲状腺结节伴钙化。胸部CT:双肺炎症,双侧胸膜增厚,前纵隔增宽。头颅MRI:双侧颞叶可见斑片状长T2信号,双侧脑室旁可见多发斑点状长T2信号,于FLAIR呈高信号(图1)。肌电图:四肢被检肌可见肌强直电位,MUP时限短,双下肢被检神经周围运动及末梢感觉传导功能未见异常。双胫神经H反射正常。皮肤活检:皮下各层结构清晰,皮下小血管较多,血管内皮细胞及外周平滑肌细胞未见异常,血管外周肥大细胞浸润。反复切片共观察7个小血管,其中3个血管是平滑肌细胞表面有典型嗜鋨颗粒(图2)。肌肉活检:骨骼肌呈肌营养不良样病理改变。大量肌纤维核内移、核聚集,少数肌纤维内肌浆块,类似病理改变可见于强直性肌营养不良(图3)。基因检测:受检者强直性肌营养不良1型DMPK基因3UTR区的CTG重复数目扩增出现两种片段:6次(正常片段)和超过50次(全突变片段),未能确定具体的CTG重复数。

注:A~C:双侧颞叶可见多发斑片状长T2信号;D~F:脑室旁可见斑点状长T2信号图1 头颅MRIFigure 1 HeadMRI
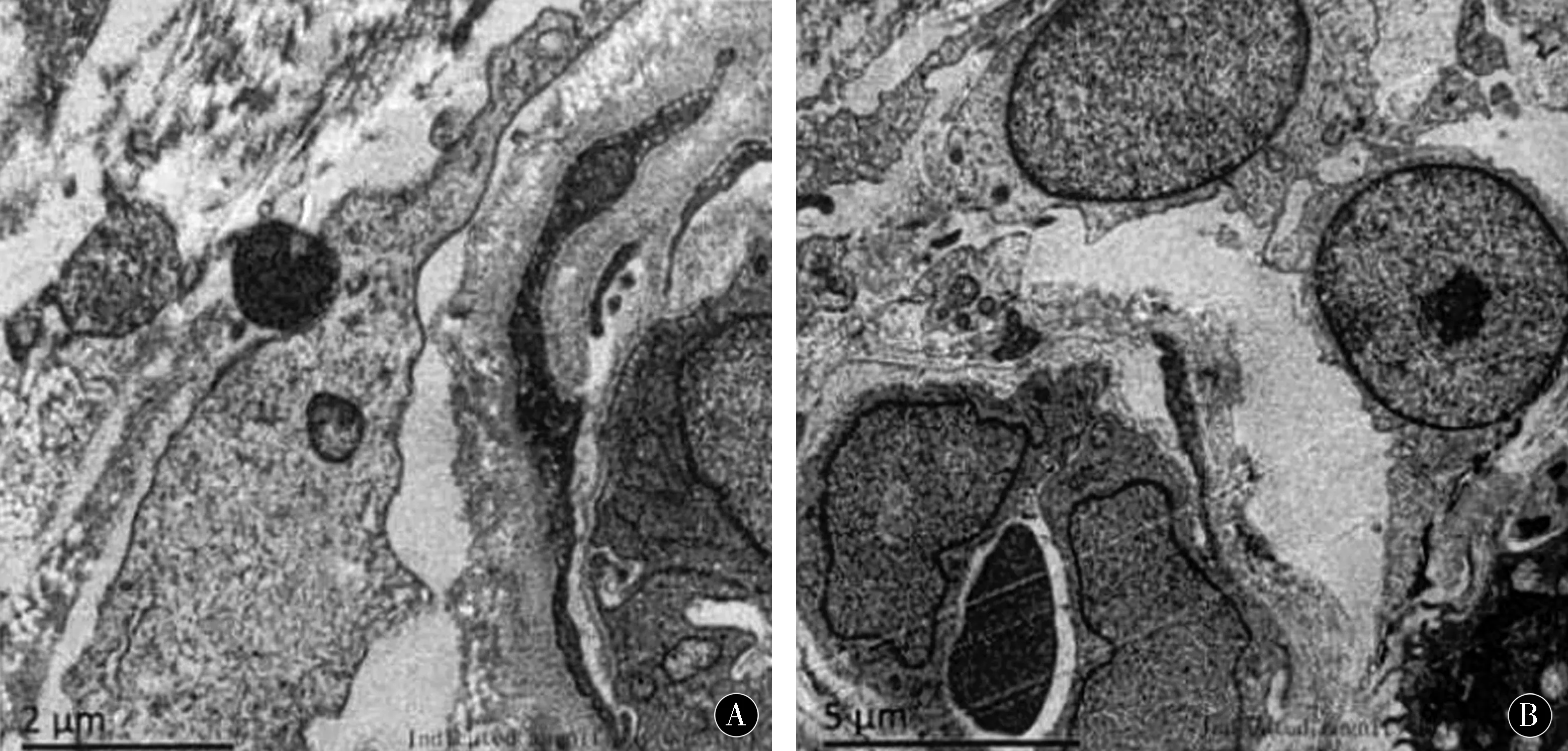
图2 皮肤活检Figure 2 Skin biopsy

注:A:HE染色:可见大量肌纤维核内移(箭头所示)、核聚集,少数肌纤维内肌浆块;B:MGT染色:可见肌浆块呈紫蓝色图3 肌肉活检Figure 3 Muscle biopsy
本例男性患者,以双眼白内障为首发症状,后出现双下肢无力的症状,同时伴有秃顶、斧状脸、高胆固醇、高脂血症,符合DM的临床表现,其确诊依赖于肌肉活检和基因检测,该患者肌肉活检可见坏死肌纤维,伴大量核内移及肌纤维膜周肌浆块,结合基因检测结果CTG重复数目扩增超过50次确诊为DM1型。
高电子密度嗜鋨颗粒沉积是诊断常染色体显性遗传性脑动脉病(cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy,CADASIL)的重要手段[1],而有关嗜锇颗粒的来源及生物化学性质尚无定论,TIKKA等[2]认为这种嗜鋨性颗粒物质或许与Notch3基因突变有关。因此,考虑本例患者可能合并Notch3基因突变,但并未争得患者同意,未做基因检测确诊。
2 讨论
强直性肌营养不良根据基因不同分为DM1型和DM2型,是成人中最常见的肌营养不良症。DM1由位于19号染色体长臂(19q13.3)的肌营养不良蛋白激酶基因(myotonic dystrophy protein kinase,DMPK)中不稳定三核苷酸即CTG的异常扩增所致;DM2由锌指蛋白9基因(zinc finger protein 9,ZNF9)的内含子中存在一个四核苷酸即CCTG的异常扩增所致[3-5]。正常人有5~37个CTG重复序列[6]; DM2由锌指蛋白9基因(zinc finger protein 9,ZNF9)的内含子中存在一个四核苷酸即CCTG的异常扩增所致[7-10]。研究表明DM1和DM2的共同临床特征涉及一种新的遗传机制,即重复RNA发挥毒性作用[11-12]。此外,DM1基因缺陷引起的选择性剪接调控失调涉及脑内三个基因:微管相关蛋白Tau(MAPT)、N-甲基D-天冬氨酸受体(NMDAR1)和淀粉样前体蛋白(APP)基因,这些基因编码蛋白表达的改变可能导致大脑参与DM1[13]。
强直性肌营养不良1型的患病率为(3~15)/10万[14],强直性肌营养不良疾病男性发病率高于女性,多在30岁以后隐匿起病,主要累及骨骼肌系统,以肌无力、肌强直和肌萎缩为特点。肌无力和肌萎缩累及头面部时,颞肌、咬肌萎缩呈“斧状脸”,颈部肌肉萎缩而稍前屈,形成“鹅颈”,累及胫前肌群时出现跨越步态及足下垂的表现。肌强直主要影响臂、手及舌的活动,用叩诊锤叩击双手大鱼际处时可见肌球。DM患者的肌张力在休息后更为明显,随着肌肉活动的“热身现象”而逐渐有所改善[15]。骨骼肌以外的临床症状与年龄密切相关,在成年患者中表现较为明显。90%的成年患者在早期出现白内障症状,可作为DM1患者早发的主要甚至唯一的临床特征为临床诊断提供重要依据[16]。呼吸系统受累引起膈肌无力和肺泡通气不足,因而导致肺部反复感染[17],呼吸衰竭是DM1患者最常见的死亡原因[18]。DM1对心脏的影响主要表现在传导系统上,尤其是心脏传导阻滞是继呼吸衰竭之后的第二大死亡原因[19]。DM1累及胃肠道时可表现为胃痛、腹泻、便秘、腹痛、大便失禁、吞咽困难、肝脏异常、肠易激综合征,其中吞咽困难是DM1患者最常见的胃肠道症状[20],肝功能异常在DM疾病中很常见,表现为丙氨酸氨基转移酶、天门冬氨酸转移酶及γ-谷氨酰转移酶升高[12,21-22]。DM1患者代谢紊乱会引起胰岛素抵抗、胆固醇升高和高甘油三酯血症[21,23]。累及内分泌系统可能会引起男性秃顶、睾丸萎缩,勃起功能障碍等,导致生育能力下降[24]。患有1型强直性肌营养不良的女性患者在怀孕期间有发生并发症的风险,包括自然流产率增加、延长分娩时间、胎盘滞留和产后出血等[25-26]。神经系统受累常见的症状包括冷漠、疲劳、认知困难、智力低下、白天嗜睡、睡眠呼吸暂停和睡眠中的周期性腿部运动[27],除了神经精神症状和认知困难外,伴糖尿病的DM1患者患脑肿瘤的风险增加,而星型细胞瘤是最常见的脑肿瘤类型[28]。
DM1颅内病变异质性较大,其影像学表现包括弥漫性脑萎缩、脑室扩大、扩张的血管周间隙、脑白质高信号及颅骨增厚等。脑白质高信号主要位于额叶和颞叶,其中前颞叶的白质病变在DM1患者中表现出较高的患病率[29],并且是1型肌营养不良的特征性表现[30]。伴脑白质病变的 DM1与CADASIL在核磁上均可表现为双侧颞极白质的病变[31],而且CADASIL 的一些特征性的核磁表现形式可出现在 DM1中,因此应当注意鉴别,本例患者头颅MRI(图1)双侧颞叶可见斑片状长T2信号,并且皮肤活检提示血管平滑肌细胞表面有典型嗜锇颗粒,因此,不排除本例患者合并有CADASIL。
DM1虽然尚无治愈方法,但积极的治疗可能会显著降低患者群体的发病率和病死率,多项研究通过对DM发病机制的进一步了解已发现许多潜在的治疗策略,包括小分子治疗、基于反义寡核苷酸(ASO)的治疗、针对被改变的DNA、RNA或下游信号通路的基因组编辑[18]、CRISPR/Cas。CRISPR/Cas系统可使细胞功能得到永久性挽救,具有DM1的巨大潜力,但是需要考虑CRISPR/Cas9引起的意外脱靶切割事件的风险[32]。RNA疗法通过靶向有毒的CUG扩张,在治疗DM1方面显示出特别的前景,而使用不同的基于寡核苷酸的方法在动物模型中取得了令人鼓舞的结果,但反义技术和RNAi的主要困难之一是缺乏有效的递送,因此,DM1疗法必须克服全功能膜的障碍才能成功地给药,一旦患者体内的传递障碍被克服,反义技术和RNA疗法有望充分发挥其良好的治疗效果[33]。这些策略在体外、细胞内和患者体内有效性和安全性目前还无确切定论,还需更深入的研究。
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26 07:17:24
中国临床医学影像杂志(2022年5期)2022-07-26 07:11:54
国际放射医学核医学杂志(2021年10期)2021-02-28 08:43:54
中华养生保健(2020年5期)2020-11-16 01:43:44
家庭科学·新健康(2019年9期)2019-10-21 03:55:48
天津农学院学报(2016年2期)2016-12-01 05:40:05
中西医结合心脑血管病杂志(2016年20期)2016-03-01 04:20:48
医学研究杂志(2015年9期)2015-07-01 17:28:00
中医研究(2014年2期)2014-03-11 20:28:18
中国中医药现代远程教育(2014年19期)2014-03-01 04:30:44
