
一步法制备高稳定性纳米α-H2Pc及其光电导性能研究*
2020-11-09 02:26韩明明张亚琴王世荣刘红丽董晓菲李祥高
功能材料 2020年10期
韩明明,刘 流,张亚琴,王世荣,刘红丽,董晓菲,李祥高
(1. 天津大学 化工学院,天津 300354; 2. 天津化学化工协同创新,天津 300072;3. 天津市功能精细化学品技术工程中心,天津 300354)
0 引 言
酞菁化合物具有非常稳定的耐酸及耐有机溶剂等特性[1]。近年来随着纳米技术和薄膜技术的应用,α-H2Pc材料表现出优异的光电转换性能,被广泛应用于光伏[2-3]、场效应晶体管[4]、光电检测器[5]、液晶显示[6]、电致发光显示[7]、电化学催化剂[8-9]等领域,并成为持续的研究热点。
H2Pc是最早被发现的酞菁化合物,有α、β和χ三种常见的晶型。β型是热力学上最稳定的晶型,α型是不稳定晶型,但其具有更优良的光电响应性能。因此,研究晶型稳定的α-H2Pc的制备条件是很重要的课题。α-H2Pc的主要制备方法有溶剂诱导[10]、球磨[11]、真空升华[12-13]、晶种合成[14-15]和热致诱导[16]等。早期Hauser等[11]采用干法和湿法球磨分别制备了α-和χ-H2Pc。但在该法的制备过程中会将球磨介质以杂质的形式带入H2Pc产物中、而且产率低,工艺条件难以控制等。S .Tamura等[13]研究了在不同合成温度下,可分别得到α-H2Pc、β-H2Pc、χ-H2Pc以及Fχ-H2Pc等产物。该方法对温度的控制要求很严苛,一般只能得到混合产物。另外,合成过程中产生的副产物和未反应的物质不易被清除,很难得到高纯度的材料。Paul J. Brach[14]采用以χ- H2Pc为晶种和一种芳香族溶剂长时间混合搅拌可以使α-H2Pc转型为χ-H2Pc。该方法重复性差。Eric B. Wasmund等[17]证明了H2Pc经过液体射流的相互作用可获得多晶型,但对溶剂的选择有很高的要求且容易得到混合产物。浦冰叶等[16]以热能为驱动力,制备出高稳定性和高光敏性的α和β-H2Pc多晶体,该方法操作简单,无繁杂的后处理工艺,但在热处理过程中容易使H2Pc原始粒子发生二次团聚,使其粒径增大,表面极性增强。通过湿法球磨中的机械力与溶剂诱导的共同作用也能制备多种晶型的H2Pc。Richard W. Radle等[18]报道在球磨条件下,芳香族、氯代烃等易使H2Pc转型为β晶型,而含有O=C基的溶剂中可制备出亚稳定晶型的H2Pc,如α-H2Pc、τ-H2Pc和χ-H2Pc,但通常为混合晶型的产物。
本文选择以硫酸为溶剂、将H2Pc的硫酸溶液滴加到由丁酮和水组成的混合溶剂中,在机械力作用下对H2Pc进行晶型调节。在不同组成比例的丁酮与水的分散及晶型调节的介质中,系统地研究了分散介质的组成、搅拌速度、晶型调节温度和时间等因素对晶型、结晶度、粒度和光敏性能等的影响,得到了高稳定性、高光电转换效率的α-H2Pc的制备方法。
1 实验部分
1.1 试剂与仪器
1.1.1 实验试剂
试剂:1,3-二亚胺基异吲哚啉(酞菁素);N,N-二甲基乙醇胺,分析纯(天津市福晨化学试剂有限公司);甲醇钠(上海阿拉丁生化科技股份有限公司);丙酮、98%硫酸、丁酮,分析纯(天津市江天化工技术有限公司);乙醇、1,2-二氯乙烷、环己酮、异丙醇,分析纯(天津市利安隆博华医药化学有限公司);聚乙烯醇缩丁醛、聚酰胺、聚碳酸酯(上海晶纯生化科技股份有限公司);N,N,N′,N′-四(4-甲基苯基)-1,1′-氨联苯二胺由本实验室合成。
测试仪器:FTIR-650红外光谱仪(天津港东科技发展股份有限公司),DelsaTMNano C 型纳米粒度仪(美国贝克曼库尔特公司),Dimension Icon型原子力显微镜(德国Bruker公司),UV2600紫外可见分光光度计(上海天美科学仪器有限公司),Turbiscan LABTM分散稳定测试仪(法国Formulaction公司),Min Flex 600 X射线粉末衍射仪(日本理学公司),SP-428静电纸分析仪测试(日本KAWAGUCHI电气株式会社,经过数字化改造)。
1.2 实 验
1.2.1 H2Pc的合成
将500 mL N,N-二甲基乙醇胺、103.4 g(0.71 mol)1,3-二亚胺基异吲哚啉和8.2 g(0.15 mol)甲醇钠加至1 L的三口瓶中。在N2保护下,加热至30 ℃使其混合物充分溶解后,继续加热至回流温度,反应液的颜色逐渐由墨绿色变为深紫色,持续反应6 h后将反应体系的温度降至100 ℃左右,趁热过滤,用乙醇和丙酮分别打浆洗涤紫色滤饼3次,干燥得到紫色固体48.5 g,收率为47.0%。
1.2.2 H2Pc的提纯与晶型调节
将2.5 g合成的H2Pc缓慢加入到50 mL剧烈搅拌的98%硫酸中,保持体系的温度在5 ℃以下,搅拌2 h使H2Pc充分溶解。然后使用分液漏斗将H2Pc浓硫酸溶液滴加到80 mL由丁酮与水组成的分散介质中,滴加完毕后,继续搅拌1 h,得到蓝色溶液,静置分层、除去上清液、过滤,分别用去离子水和甲醇洗涤,直至滤液的电导率<2 μS/cm。最后将滤饼真空冷冻干燥得到1.9 g的H2Pc,收率76.0%。采用X射线粉末衍射表征不同条件下得到的样品的晶型。
研究不同组成分散介质、搅拌速度、晶型调节温度和时间等条件对晶型转化的影响。
1.2.3 纳米H2Pc粒子分散液的配制
将1∶1∶500(W/W)的聚乙烯醇缩丁醛(M.W:90000~120000)、H2Pc纳米粒子和直径为1 mm的锆珠加至球磨罐内,再加入1∶1(V/V)的丁酮和环己酮的混合溶剂。以360 r/min的转速,在对辊机上球磨分散1、2 、3 和4 h,然后用孔径为10 μm聚丙烯膜过滤,得到蓝色α-H2Pc/MEK/CYC/PVB分散液。
为了分析制备的α-H2Pc的晶型稳定性,将其冷冻干燥得到蓝色粉末状样品,用X射线粉末衍射表征获得样品的晶型。
1.2.4 纳米α-H2Pc的光电导性能
将含4%聚酰胺的甲醇溶液涂布在镜面铝箔上制备预涂层;按照上述1.2.2方法制备的H2Pc作为载流子产生材料,与聚乙烯醇缩丁醛树脂(质量比为1∶1)混合分散在体积比为1∶1的丁酮和环己酮溶液中制备成分散液,涂布在预涂层之上,干燥后得到载流子产生层;将以质量比为1∶1的聚碳酸酯与N,N,N′,N′-四(4-甲基苯基)氨联苯二胺混合溶解于1,2-二氯乙烷和甲苯的混合液中制备固含量为16%(W/W)的空穴传输材料溶液,涂布在载流子产生层之上,干燥后得到多层光电导器件。
分别测试充电电位(V0)、残余电位(Vr)、暗衰率(Rd)和半衰减曝光量(E1/2),来分析讨论材料的光电导性能。
2 结果与讨论
具有光电响应的纳米有机颜料的经典纯化和晶型调节分别为酸糊溶解并在低温的水中析出,获得洗涤干净的材料,将其在合适的分散介质中调节晶型,制备所需要的纳米晶型产物。在该过程中,当酞菁材料的硫酸溶液滴加到水中时,即使剧烈搅拌也不可避免地会产生局部过热而导致酞菁环的裂解进而产生杂质并降低收率。另外,必须反复过滤、洗涤,再转移至适当的介质体系中调节晶型,工艺流程长、效率低下。本文将水和晶型调节剂丁酮的混合体系在强力搅拌下形成W/O型乳液,当H2Pc样品的硫酸溶液滴加到分散体系中时,先进入丁酮连续相,被瞬间分散成微滴后再进入水相,并从水相中析出,避免瞬时大量放热的问题,之后形成的纳米H2Pc粒子反复经过水/丁酮界面诱导的作用下,转化为高结晶度的α-H2Pc纳米。这方法具有简便、不需要低温环境、快捷的特点。制备流程见图1。
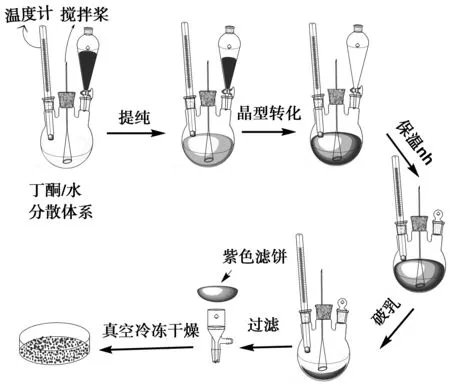
图1 一步法制备α-H2Pc纳米粒子的流程图
2.1 分散介质对H2Pc晶型的影响
分散介质中的溶剂分子可以影响H2Pc分子的堆积方式,因此对晶型的转化程度以及粒径的大小有一定的影响[19]。为了研究溶剂组成对H2Pc转化程度的影响,考虑到丁酮在水中的质量百分比在26.8%~88.2%之间才能形成分散体系,以及尽量多使用水的原则,选择体积比分别为1∶0、1∶1和1∶3的丁酮/水(丁酮的质量分数分别为44.6%和21.2%)作为分散介质对合成的H2Pc进行晶型调节。分别向两种分散体系的水相中加入微量的NaCl,测试分散体系的电导率与搅拌速度的关系,见图2。其中曲线(a)为室温和(b)40 ℃, 1∶3, 1.25 mg/L NaCl,曲线 (c)为室温和(d)40 ℃, 1∶1, 12.5 mg/L NaCl。从图中曲线可见,无论在室温还是40 ℃,丁酮与水的体积比为1∶1时,在200~1 000 r/min搅拌速度下可观察到该体系呈乳白色的分散状态,如图3(a),同时电导率的变化清楚地表明该体系形成了O/W类型乳液;而当丁酮与水为1∶3时,初期形成的是互溶体系,但搅拌速度在300~600 r/min之间,体系虽然仍处于透明的状态,如图3(b),但电导率明显降低,表明此时形成了W/O类型的分散体系。当搅拌速度达到700~1 000 r/min时,电导率则快速上升,同时可以观察到体系转变为乳白色,如图3(c),说明分散体系在这个条件下转化成了O/W型乳液。这个现象还说明在一定的搅拌速度下,丁酮在水中的溶解度范围内,均相溶液可以转变成不同形式的分散体系。
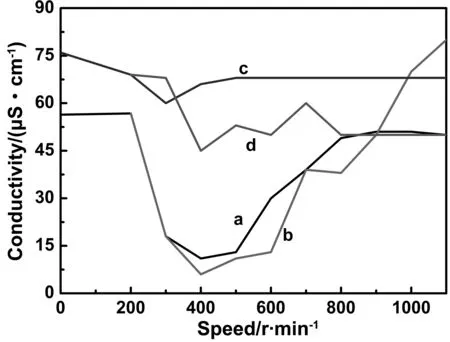
图2 丁酮/NaCl水溶液分散体系的电导率与搅拌速度的关系

图3 不同搅拌速度下丁酮/水体系的溶液状态
在40 ℃、搅拌速度为400 r/min、调节晶型1 h的条件下,按照1.2.2的方法制备得到不同分散体系中的H2Pc产物。可以观察到丁酮/水体积比为1∶1时的分散介质呈乳白色,H2Pc硫酸溶液滴加到其中时放热明显;而介质完全为丁酮时,混合体系呈墨绿色溶液的状态且没有沉淀析出,说明H2Pc硫酸溶液与丁酮是混溶的,向其中慢慢滴加等体积的水时,析出蓝色沉淀;向丁酮/水体积比为1∶3的分散介质中滴加H2Pc硫酸溶液时,观察到体系温度没有产生变化,而且H2Pc立即析出使体系呈蓝色,说明分散体系形成W/O型的乳液。3种不同体系中制备的H2Pc样品的X射线衍射谱见图4,根据MDJ Jade 6.0软件计算出不同组成比例的结晶度。从图4可以看出,3种H2Pc产物都属于α晶型(2θ=6.8、7.4、13.6、14.9、15.9、16.7、24.8、26.3、27.9°)[20]。但丁酮/水的比例为1∶3时,其α晶型的特征衍射峰强度增加,结晶度最高为91.6%,在体积比为1∶1分散介质中制备的样品的结晶度为90.3%,用水稀释丁酮溶液析出的蓝色粉末是结晶度为85.4%的α-H2Pc。表明在水与丁酮组成的分散体系中,丁酮可以高效地诱导α-H2Pc的生成。尤其是丁酮/水的比例为1∶3时,能够形成W/O型分散介质,既能获得高结晶度的产物,又符合绿色制备的理念。
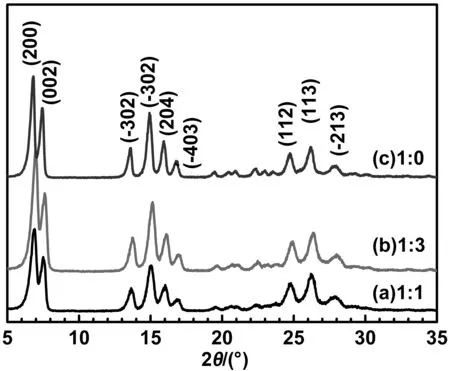
图4 不同组成比例的丁酮/水为分散介质制备的H2Pc的XRD谱
2.2 搅拌速度对H2Pc晶型的影响
在分散体系中,纳米α-H2Pc粒子的制备过程必须经过两个阶段。首先,当H2Pc的硫酸溶液滴加到丁酮/水分散介质时,首先与形成W/O乳液中的连续相丁酮接触,由于硫酸具有强亲水性,然后快速经过油/水界面进入水微滴中,立即析出并在丁酮的作用下直接诱导形成α型晶核。在2.1中分散介质只有丁酮时,硫酸溶液中的H2Pc不能析出,而与丁酮互溶,同时在水中析出时只能得到无定型H2Pc,因此α-H2Pc晶核的形成是发生在丁酮与水的界面上。当这一过程持续进行时,随着分散体系的组成或调节条件的不同,当连续相中的硫酸微滴进入分散相时,会影响H2Pc分子的析出速度以及沿晶核的堆积生长速度。体系的搅拌速度对晶体的生长以及纳米α-H2Pc粒子的粒径大小有着重要的影响。图5是以1:3的丁酮/水为分散体系,在40 ℃,搅拌速度分别为400、500和600 r/min,晶型调节5 h时得到的α-H2Pc的X射线衍射图。
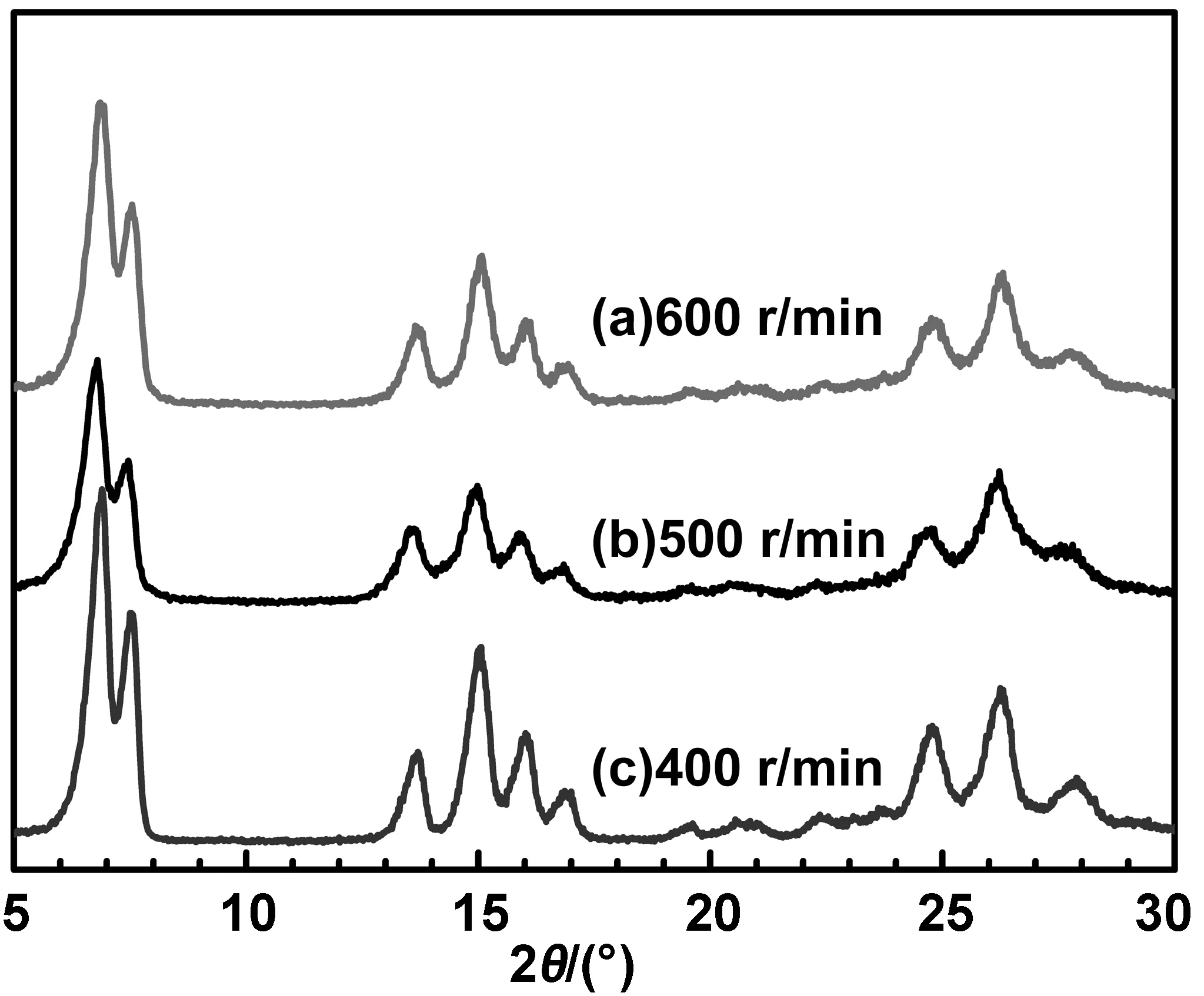
图5 温度40 ℃时不同搅拌速度下转型H2Pc的XRD谱图
从图中的XRD谱可看出,与搅拌速度为400 r/min相比,500和600 r/min下制备的α-H2Pc的特征衍射峰强度稍微减弱,2θ= 27.8°处的衍射峰强度明显减弱,说明搅拌速度的提高对(-213)晶面的生长影响较大。因此通过搅拌作用产生的剪切力会削弱H2Pc的结晶状态[21]。晶型转变发生在相转移的过程中,因此H2Pc分子可能发生堆积缺陷而影响晶体的生长,使个别衍射峰强减弱,结晶度降低。另一方面,搅拌速率越大,剪切速率就越大,从水滴中析出的微粒就越小。体系破乳后,不同搅拌速度下产物的粒度分布,如图6所示。明显可见,搅拌速度在400 r/min时,虽然H2Pc的平均粒径为72.5 nm,但其粒径分布宽达100 nm,在40~140 nm之间;提高到500 r/min时,粒径分布宽度显著缩小在100~134 nm之间的34 nm,但其平均粒径最大,为115.7 nm;当搅拌速度达到600 r/min时,不仅平均粒径减小至68.8 nm,而且分布也进一步收窄在57~87 nm之间的30 nm。3种样品的结晶度分别为91.6%、90.7%和90.2%。显然,以体积比为1∶3的丁酮/水为分散体系,在403 ℃,晶型调节时间为5 h的条件下,搅拌速度的提高,可有效的减小α-H2Pc纳米粒子的粒径分布,但不一定使其平均粒径减小,只有当搅拌速度调节到合适的程度才能获得结晶度高、平均粒径小、而且分布窄的纳米α-H2Pc产物。
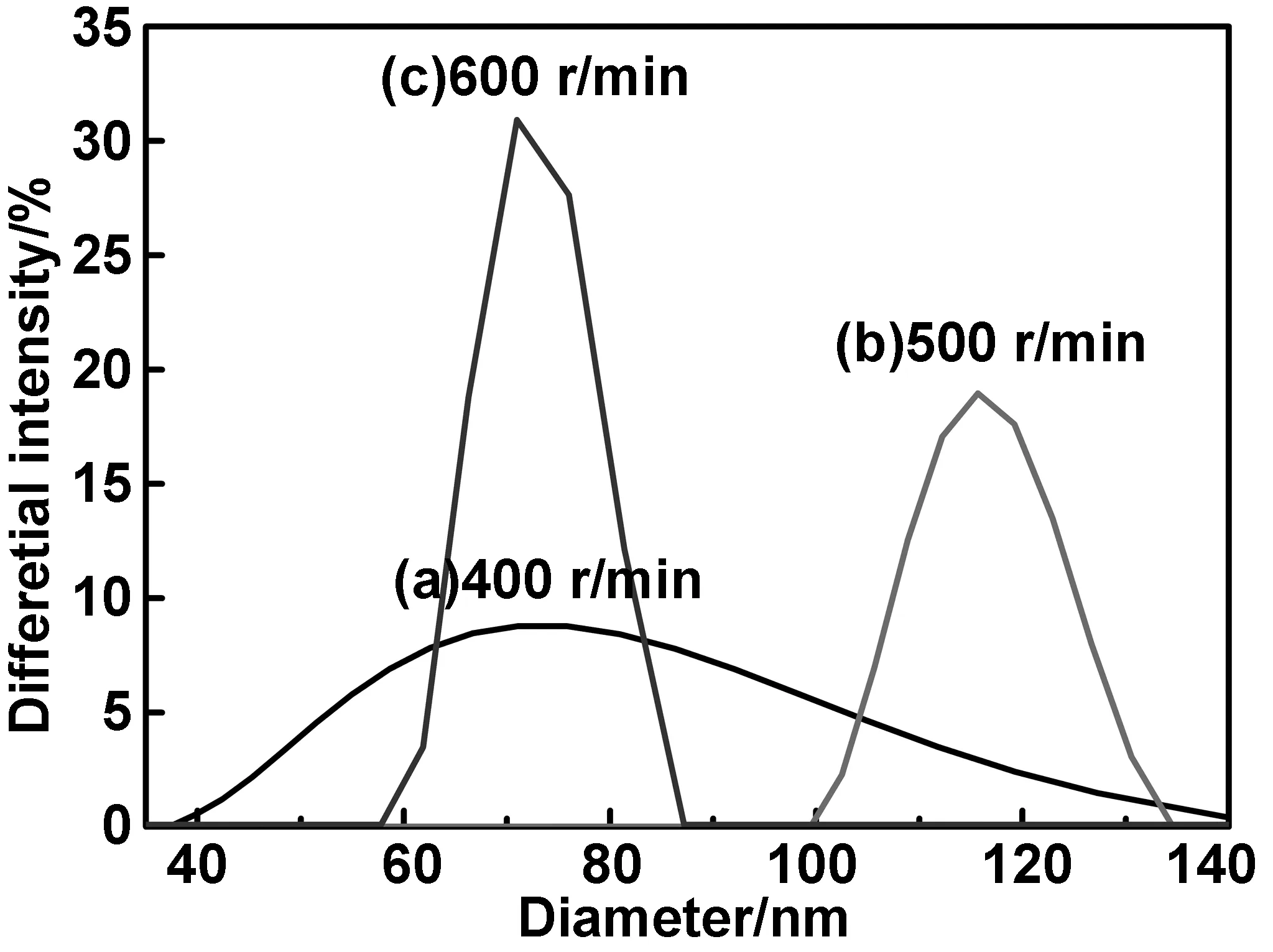
图6 不同搅拌速度下转型 H2Pc的粒径分布
2.3 晶型调节温度对H2Pc晶型的影响
以体积比为 1∶3 的丁酮/水作为分散介质,研究晶型调节温度对H2Pc的晶型转变的影响。在600 r/min、晶型调节时间为1 h时,在-10~50 ℃范围内的不同温度下制备的H2Pc的X射线衍射谱见图7,计算其结晶度数据见表1。由图7可以看出,转型温度<0 ℃时,得到的H2Pc粉末基本是无定型,存在不明显的α晶系(6.8、7.4、13.6、14.9、15.9、16.7、24.8、26.3、27.9°)的特征衍射峰。此时,α-H2Pc的含量很低,且继续降低体系温度也不会有明显改善,只有当体系温度升高至20 ℃时,α-H2Pc的比例才呈现明显增加的趋势,30 ℃后趋于平稳。

图7 不同温度下转型H2Pc的XRD图

表1 不同温度下转型H2Pc的结晶度数据
这说明在丁酮/水分散体系中,在H2SO4的存在下,以热力学的角度,α-H2Pc是更稳定的形式。H2Pc分子在进入丁酮相时从无序堆积快速向α晶型堆积转化。其晶胞堆积和纳米粒子的AFM照片见图8,可以看出该方法制备的α-H2Pc的形貌呈棒状结构。

图8 α-H2Pc的晶胞堆积和AFM图
图9是一步法以40 ℃转型的α-H2Pc和无定型H2Pc的红外吸收光谱图。振动吸收的变化是由于不同晶型的H2Pc中相邻分子的排列方向不同而引起的,是区分不同晶型H2Pc的主要特征之一。在3 300 cm-1左右的N-H弯曲振动吸收也会因为H2Pc晶型的不同而产生差[22-23]。浦冰叶[16]测得提纯后的H2Pc在715.4 cm-1和765.6 cm-1以及3 293.8 cm-1附近处出现红外吸收峰,见图9(a),而从图9(b)中的局部吸收谱可看出,α-H2Pc在同样的峰位置处产生吸收。由此判定两者为同种晶型均为α-H2Pc,结合X射线衍射的分析结论,证明在40 ℃的温度下转型H2Pc得到的是α晶型。
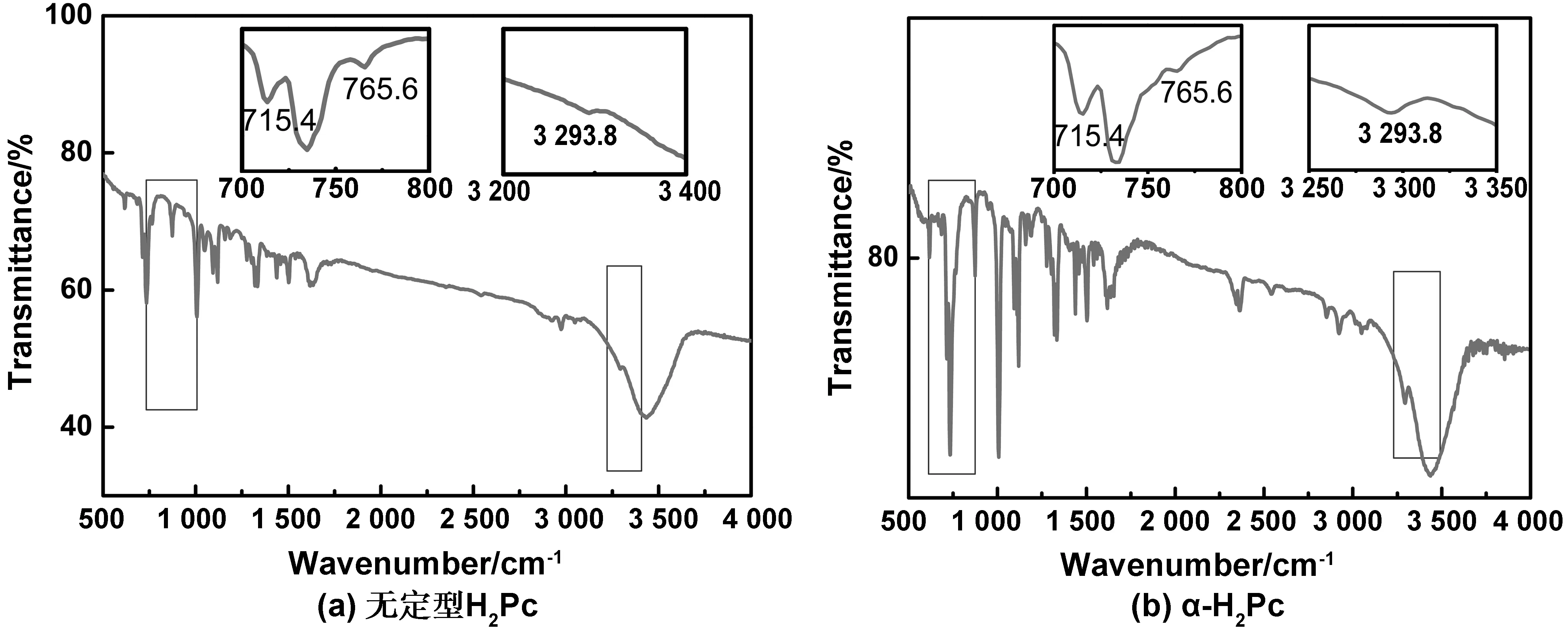
图9 H2Pc的红外吸收光谱
2.4 晶型调节时间对H2Pc晶型的影响
晶型调节时间也可能对H2Pc的晶型转化程度有一定的影响。在丁酮/水体积比为1∶3的分散体系、40 ℃和搅拌速度为600 r/min下,分别调节晶型1、3、5、7和8 h,得到产物的X射线衍射谱如图10,计算得到的结晶度数据如表2,从图10中可见,在40 ℃下连续晶型调节5 h以内,时间越长,α-H2Pc的特征衍射峰强越强,峰型越尖锐,当增加至7 h时,开始出现2θ=8.9°的特征衍射峰,这是典型的β-H2Pc特征衍射峰。说明晶型调节时间过长,H2Pc则会由α晶型向β晶型转变。
由表2可见,若晶型调节时间在5 h之内,随着时间的增加,α-H2Pc的结晶度也随之增长。在5 h时,α-H2Pc的结晶度达到最高的92.90%,当增加至7 h时,其结晶度稍微有所下降。
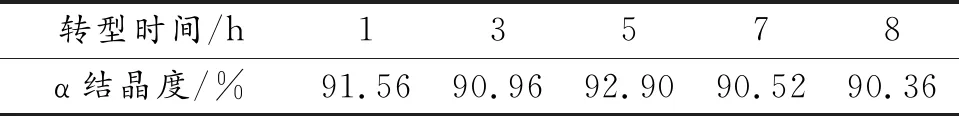
表2 温度40 ℃时不同转型时间下转型H2Pc的结晶度的数据
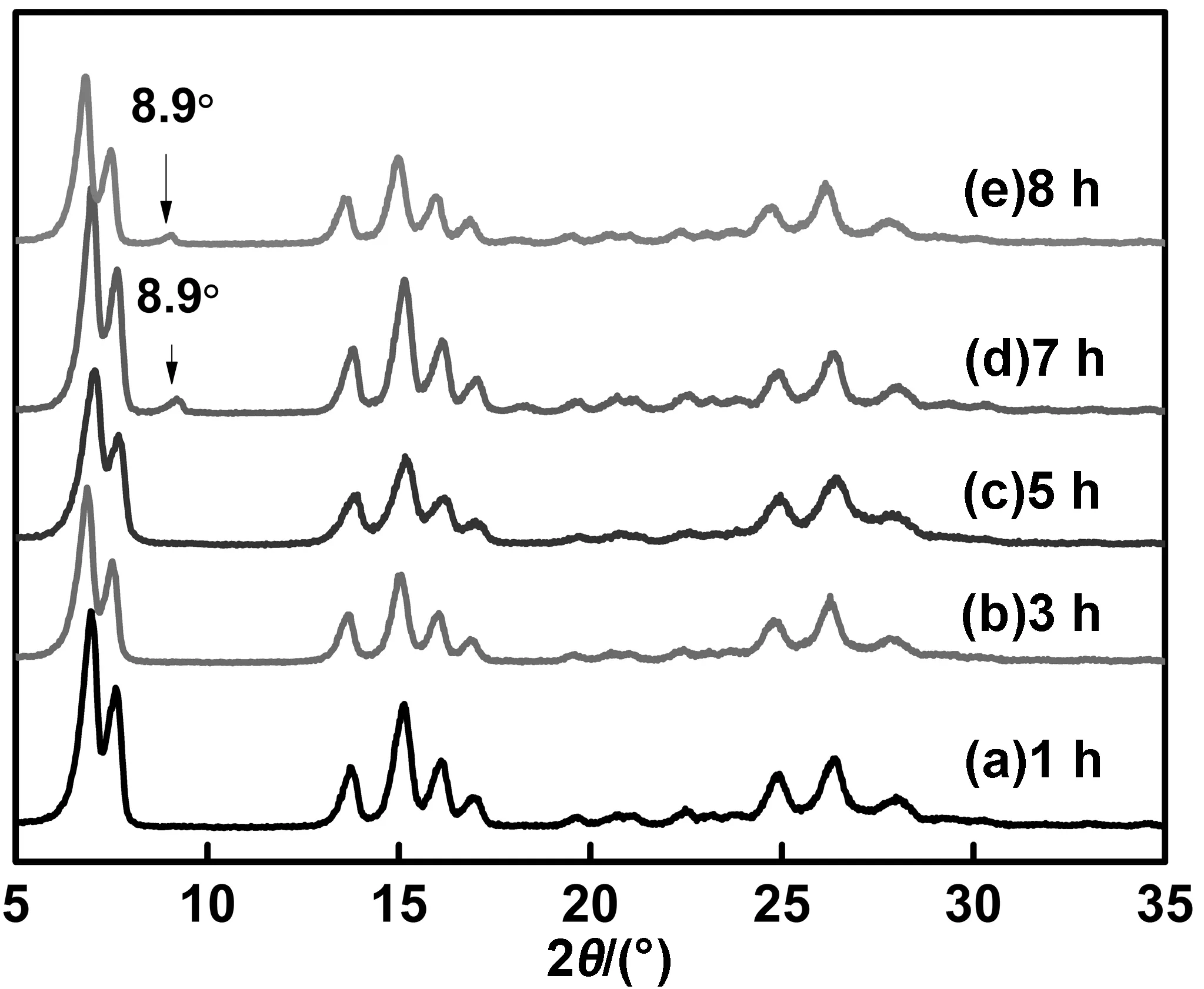
图10 温度40 ℃时不同转型时间下转型H2Pc的XRD谱图
2.5 H2Pc晶型稳定性的影响
H2Pc属于同质多晶型有机晶体,在机械力的作用下易转变为能量低、结构稳定但光电响应差的β-H2Pc。因为在制备激光有机光电导器件时,需将制备的、α-H2Pc树脂和有机溶剂混合在一起并通过球磨制备载流子产生材料分散液。所以球磨时保证分散液中α-H2Pc的晶型稳定显得尤为重要。
从图11中可看出,丁酮/水为分散介质制备的α-H2Pc在球磨4 h之内晶型稳定性良好,没有其它种类晶型的H2Pc出现,并随着球磨时间的增加, α-H2Pc的特征衍射峰峰强增加。因此,采用一步法制备的α-H2Pc纳米粒子工艺中,能够保证丁酮/水充分润湿H2Pc纳米粒子,使无定型H2Pc发生相转变,得到高结晶度和高晶型稳定性的α-H2Pc纳米粒子。
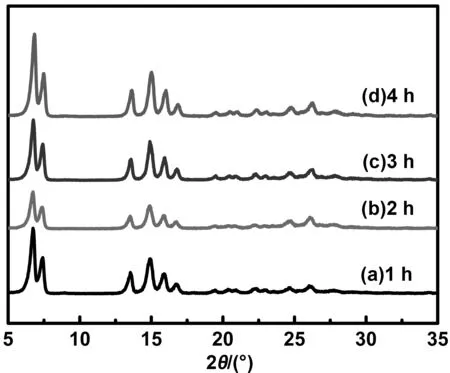
图11 H2Pc的晶型稳定性XRD谱图
以丁酮/水的体积比为1∶3的体系,在40 ℃,搅拌速度为600 r/min的条件下,选择晶型调节时间分别为3、5和7 h得到的纳米α-H2Pc在MEK/CYC/PVB的分散液中的紫外-可见吸收光谱如图12所示,该条件下制备的α-H2Pc在600~900 nm范围内有良好的吸收,并且酞菁化合物在紫外-可见光的范围内产生两个吸收带:分别在300~400 nm(B带)和600~900 nm(Q带)的两段区域内[24]。但该方法制备的α-H2Pc纳米粒子分别在610和690 nm附近有两个吸收带,说明在600~900 nm范围内的吸收峰发生了“Davydov”裂分,一个发生了红移,另一个发生了蓝移[25-26]。从图12可知,增加晶型调节时间,其H2Pc样品的最大吸收峰的位置没有变化,但其强度增加,由表2可知,晶型调节5 h时,结晶度最高,H2Pc分子的排列最为规整有序,表明本文建立的方法可以制备高结晶度和高稳定性的α-H2Pc。
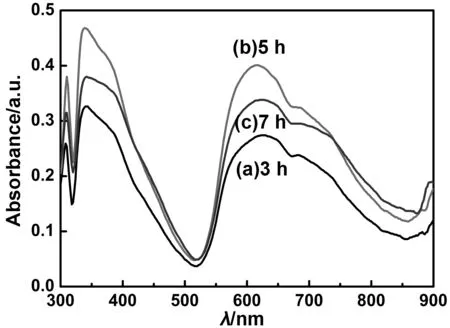
图12 不同晶型调节时间下制备的H2Pc在分散液中的紫外-可见吸收光谱
一步法制备α-H2Pc纳米粒子的过程中晶型转变发生在H2Pc由丁酮相被萃取至水相的界面诱导而导致H2Pc相转移,每个晶核的形成都是经过丁酮/水界面,因此该方法能够完全润湿制备的α-H2Pc纳米粒子。而传统方法需经过提纯、洗涤和干燥步骤,干燥时H2Pc粒子会发生聚集,从而使部分H2Pc在晶型调节过程中不能完全被转型溶剂润湿,导致转型不完全,晶型稳定性较差。
2.6 H2Pc光电导性能的影响
以体积比为1∶3的丁酮/水为分散介质,在40 ℃,晶型调节时间5 h,搅拌速度分别为400 、500 和600 r/min的条件下制备的α-H2Pc纳米粒子,通过配方配制分散液并测试其24天的透过率,并计算其透过率的变化率分别为6.80%、1.39%和1.26%。如图13所示,当搅拌速度为600 r/min时,其24天透过率的变化率最小,说明在此搅拌速度下制备的α-H2Pc纳米粒子最稳定,其分散液用DelsaTMNano C Particle Analyzer测试其zeta电位为-48.9 mV。通过上述zeta电位数值和透过率变化率可以证明,搅拌速度为600 r/min时制备的α-H2Pc纳米粒子在聚乙烯醇缩丁醛的丁酮/环己酮溶液中的分散稳定性最好。
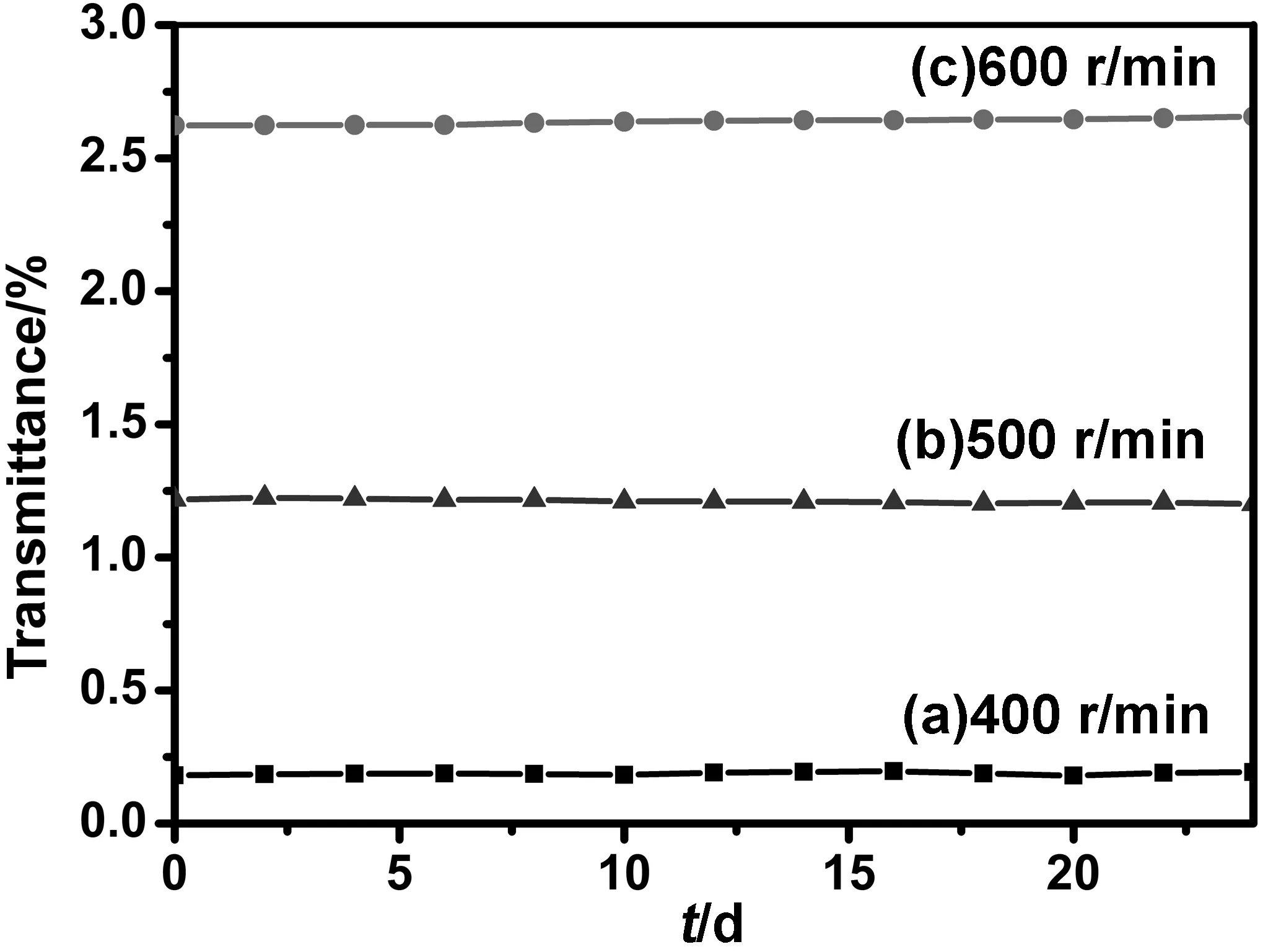
图13 不同搅拌速度下制备的α-H2Pc纳米粒子的分散液透过率随时间的变化曲线
为了研究制备的纳米α-H2Pc的光电导性能,设计结构为Al/UCL/CGL/HTL的激光光电导器件。以体积比为1∶3的丁酮/水为分散介质,在40 ℃,晶型调节时间5 h,搅拌速度分别为400、500和 600 r/min的条件下制备的α-H2Pc纳米粒子作为载流子产生材料制备激光有机光导器件,测试其光电导性能如下:
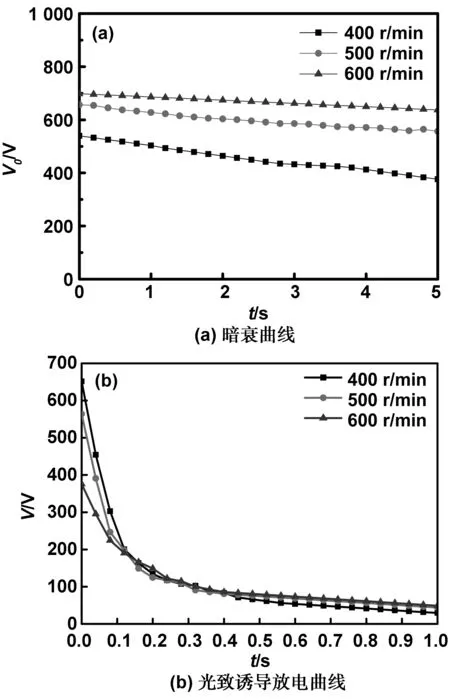
图14 不同搅拌速度条件下获得的H2Pc作为电荷产生材料制备的OPC的暗衰曲线和光致诱导放电曲线

表3 不同搅拌速度下制备的α-H2Pc作为电荷产生材料制备的OPC的光敏数据
由图14不同搅拌速度下获得的α-H2Pc作为电荷产生材料制备OPC的暗衰曲线(a)和光致诱导放电曲线(b)和表3 OPC的光敏数据可看出,当提高搅拌速度时,其充电电位(V0)升高,暗衰率(Rd)降低,残余电位(V0)降低,光敏性(E1/2)显著变化。说明该条件下制备的α-H2Pc作为载流子产生材料制备的器件接受和保持电荷的能力随着搅拌速度的提高而增强,光电导性也同样与搅拌速度呈正相关,并且表明600 r/min的搅拌速度下制备的α-H2Pc的光电导性能最好。由图6可见,搅拌速度增大,α-H2Pc的纳米粒径减小,这是由于粒径的大小对光敏性有影响[27]。α-H2Pc的纳米粒径与其表面积有关,粒径越小,其表面积越大,从而提高了α-H2Pc产生载流子的数目。因此搅拌速度越大,其光敏性能越好。结果表明,以体积比1∶3的丁酮/水为分散介质,600 r/min的搅拌速度、40 ℃和晶型调节5 h的条件下制备的α-H2Pc作为载流子产生材料时,具有良好的光电导性能。
3 结 论
通过本文的研究可以得到以下结论:
(1)在丁酮与水的比例为1∶3(V/V)、搅拌速度为600 r/min、晶型调节温度为40 ℃、时间为5 h的条件下,将H2Pc的浓硫酸溶液滴加到分散乳液中,可以方便地一步法制 备α-H2Pc,具有简便,节约时间和成本、防止 H2Pc 转型的二次团聚的特点。
(2)晶型调节温度对α-H2Pc的晶型转化影响较大。在40 ℃,连续分散5 h下,可以获得结晶度为92.9%的α-H2Pc产物;在0 ℃以下其产物为无定型H2Pc。
(3)制备的α-H2Pc具有优异的晶型稳定性能,以其作为载流子产生材料制备的激光有机光电导器件显示了良好的光电导性能。
猜你喜欢
弹性体(2021年6期)2021-02-12
陶瓷学报(2020年2期)2020-10-27
化学工程师(2020年5期)2020-06-30
生物工程学报(2018年5期)2018-06-11
林产化学与工业(2017年5期)2017-11-07
河南化工(2017年7期)2017-08-12
中国塑料(2015年6期)2015-11-13
中国塑料(2015年8期)2015-10-14
江苏农业科学(2015年1期)2015-04-17
科技创新导报(2014年34期)2015-01-13
