
泊那替尼固体脂质纳米粒的制备与质量评价
2020-10-29 13:47熊远果沈瑶张洪
医药导报 2020年11期
熊远果,沈瑶,张洪
(武汉大学人民医院药学部,武汉 430060 )
泊那替尼(ponatinib)是第3代酪氨酸酶抑制药(tyrosine protein kinase inhibitors,TKI),可用于对第1代(伊马替尼)和第2代(达沙替尼)TKI耐药的慢性粒细胞白血病(chronic myelocytic leukemia,CML)的治疗[1-2]。目前,泊那替尼主要用于治疗两种耐药型白血病,即CML和费城染色体阳性急性淋巴细胞白血病[Philadelphia chromosome-positive(Ph+) acute lymphoblastic leukemia,Ph+ALL]成年患者,并可用于对加速期耐药性白血病的治疗[3-4]。泊那替尼对突变的费城染色体上BCR-ABL融合基因有明显抑制作用,并可直接抑制T315I突变,延长耐药患者生存周期,提高患者生活质量[5-6]。X-射线晶体分析表明,泊那替尼以范德华力与T315I突变残留结合,抑制突变蛋白表达[7-8]。但泊那替尼水溶性较差,上市片剂主要吸收部位为胃部,生物利用度低,不良反应发生率高,制约了该药在临床上的使用[9]。
固体脂质纳米粒(solid lipid nanoparticles,SLN)是近年发展的一种新型毫微粒类给药系统,以固态的天然或合成的类脂材料将药物包裹于类脂核中制成粒径50~1000 nm的固态胶粒给药体系[10]。SLN作为用于控制药物释放的新型纳米胶团载体的给药系统,既具备聚合物纳米粒稳定性良好、药物缓控释的优势,又兼具脂质体、乳剂低毒性、生理相容性好、良好靶向性以及可大规模生产等优点,并且药物包封于固体脂质中,可避免化学或酶降解,增加药物稳定性[11-13]。SLN可用于口服给药、经皮给药以及静脉给药等多种给药途径,应用前景广泛[14-15]。
为促进泊那替尼在临床的使用,使其更安全、有效地作用于临床,笔者采用乳化蒸发-低温固化法制备口服给药的泊那替尼固体脂质纳米粒(P-SLN)混悬液。以平均粒径(size)、多聚分散指数(polymer dispersity index,PDI)以及包封率(entrapment efficiency,EE)对其制备工艺和处方进行优化,并对其质量结构和理化性质进行初步探讨,以期为开发泊那替尼新剂型提供实验基础和理论依据。
1 仪器与试药
1.1仪器 Agilent 1100型高效液相色谱仪(DAD检测器,美国Agilent公司);UV-1800型紫外-可见分光光度计(日本岛津公司);TG328A型电子分析天平(上海天平仪器厂,感量:0.000 1 g);JEM-2100 (HR)高分辨透射电镜(日本Jeol公司);Zetasizer ZS90型激光粒度仪(英国Malvern公司)。
1.2试药 泊那替尼(武汉星耀艾克生物医药有限责任公司,含量:99.0%,批号:06771);泊那替尼对照品(上海翰香生化制品有限公司,含量:99.3%,批号:20121226);泊洛沙姆188(武汉国奥联信生物科技有限公司,批号:C5H1002);蛋黄卵磷脂(国药集团化学试剂有限公司,批号:69014933);单硬脂酸甘油酯(国药集团化学试剂有限公司,批号:30093226);甲醇[美国天地公司,高效液相色谱(HPLC)纯];乙腈(美国天地公司,HPLC纯);水为超纯水(本实验室自制),其他试剂均为分析纯。
2 方法与结果
2.1P-SLN的制备 精密称取处方量泊那替尼10 mg、单硬脂酸甘油酯100 mg、蛋黄卵磷脂75 mg,溶于适量无水乙醇,(75±2) ℃水浴形成有机相。另取100 mg泊洛沙姆188溶于相同温度水,构成水相。将有机相缓慢加入水相,高速乳化10 min(10 000 r·min-1)。乳化后,等温下继续搅拌15 min(1000 r·min-1),除去有机溶剂。将所得半透明体系快速分散于0~2 ℃水相,高速均质10 min(10 000 r·min-1),即得P-SLN混悬液。同时,在空白SLN制备的方法中,除有机相中不加入泊那替尼外,其余与制备P-SLN方法相同。
2.2含量测定
2.2.1检测波长选择 精密称取泊那替尼对照品10 mg,置100 mL量瓶,并用无水乙醇定容,摇匀,即得100 μg·mL-1泊那替尼浓贮备液。以浓贮备液配制15 μg·mL-1泊那替尼标准溶液,以空白乙醇溶液作参比,利用紫外-可见分光光度计,于190~800 nm全波长进行扫描,结果显示泊那替尼有多个吸收峰,考虑到辅料在低波长范围内有干扰作用,选择280 nm为检测波长。
2.2.2色谱条件 根据前期预实验及文献[16-17],确定色谱柱:Inertsil ODS-3柱(250 mm×4.6 mm,5 μm);流动相:乙腈-0.2%三乙胺乙酸缓冲盐(85:15,pH值=7);检测波长:280 nm;流速:1.0 mL·min-1;柱温:30 ℃;进样量:20 μL。
2.2.3专属性考察 按处方比例制备空白SLN,加入无水乙醇溶解,过孔径0.45 μm膜进样测定。同时按照处方比例制备含药SLN,同法进行测定,其HPLC图见图1(测定浓度10 μg·mL-1)。由图1可知SLN中辅料对样品含量测定干扰较小,说明该方法专属性好。
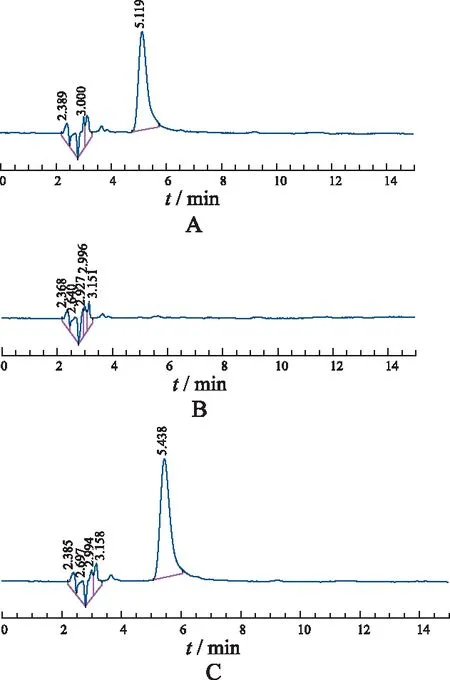
A.泊那替尼对照品;B.空白固体脂质纳米粒;C.P-SLN样品。
Fig.1HPLCchromatogramofP-SLN
2.2.4线性范围考察 标准溶液的制备:精密称量泊那替尼对照品10 mg,置100 mL量瓶,加无水乙醇溶解,摇匀,定容至刻度,即得泊那替尼对照品溶液。
标准曲线的绘制:分别精密量取上述泊那替尼对照品标准溶液4.0,3.5,3.0,2.5,2.0,1.5,1.0,0.5,0.1 mL于10 mL量瓶,加无水乙醇至刻度。配成浓度分别为40,35,30,25,20,15,10,5,1 μg·mL-1系列标准溶液,按“2.2.2”项方法进样,依次进样量20 μL。每个样品溶液进样5次,记录色谱图,计算峰面积。以5次测定的泊那替尼峰面积平均值(A)对进样浓度(μg·mL-1)做线性回归,结果为:A=33.17C+9.286 7 (r=0.999 7) (1~40 μg·mL-1)(n=5)。结果表明,泊那替尼在1~40 μg·mL-1范围内样品浓度与峰面积呈良好的线性关系。
2.2.5精密度和回收率实验 精密度测定:取“2.2.4”项泊那替尼对照品溶液,分别精密配制高、中、低(30,20,10 μg·mL-1)浓度溶液,1 d内连续进样5次,不同天内,每天进样1次,连续5 d,进行测定。结果日内精密度RSD分别为0.14%,0.28%,0.16%,日间精密度RSD为0.67%,0.49%,1.49%。
回收率测定:取“2.2.4”项泊那替尼对照品溶液,分别精密配制高、中、低(30,20,10 μg·mL-1)浓度溶液。精密量取空白SLN混悬液1.0 mL,共3份,分别准确加入上述对照品溶液1.0 mL,过滤,按“2.2.2”项进样。结果回收率分别为(99.25±0.05)%、(101.59±0.10)%、(100.28±0.08)%。
2.3包封率的测定 精密移取P-SLN混悬液1.0 mL,于4 ℃下高速离心30 min(12 000 r·min-1),去除上清液,沉淀用 0.1 mol·L-1盐酸溶液清洗3次,除去未包封的药物,加入乙腈1.0 mL和蒸馏水1.0 mL破乳,得到样品A;另精密量取P-SLN混悬液1.0 mL,直接加乙腈破乳1 mL,加蒸馏水至与A液等体积,得样品B。HPLC法测定A、B两样品中泊那替尼含量,按以下公式计算包封率,包封率(EE,%)=(样品A中药物含量/样品B中药物含量)×100%。
2.4单因素实验 根据预实验结果,制备工艺条件是影响固体脂质纳米粒粒径和PDI的主要因素。因此,本实验对制备工艺条件作了进一步的考察和优化:固化方法(自然固化、冰水浴固化、机械搅拌固化、高速均质固化)、纳米乳与冰水浴(0~2 ℃)体积比(1:1~1:20)、乳化及固化均质时间(5~15 min、2~20 min)、乳化温度(65~85 ℃)。结果见表1~4。根据粒径和PDI确定最优制备工艺为:冰水浴高速均质固化,纳米乳与冰水浴体积比为1:5,乳化及固化均质时间均为10 min,乳化温度为75 ℃。在此制备工艺下,所制纳米粒粒径较小,粒径分布比较均匀。

表1 不同固化方式对P-SLN粒径和PDI的影响
2.5正交设计实验 根据单因素实验可知,各种乳化剂比例、载体材料用量和投药量对P-SLN的各项指标参数有较大影响。因此在预实验和单因素考察的基础上,选择泊那替尼(A)、单硬脂酸甘油酯(B)、蛋黄卵磷脂(C)和泊洛沙姆188(D)用量4个因素,各因素设计3个水平,按正交设计的原理进行实验,考察因素水平见表5,正交实验结果见表6,方差分析结果见表7。
表2 纳米乳与冰水浴体积比对P-SLN粒径和PDI的影响
表3 温度对P-SLN粒径和PDI的影响
表4 高速均质时间对P-SLN粒径和PDI的影响

表5 正交实验因素水平表

表6 L9(34)正交实验方案及结果

表7 方差分析结果
为全面综合评价P-SLN制备工艺,本实验采用DF来进行综合评分[18-19]。其中,评价指标(di)包括P-SLN粒径(d1)、PDI(d2)和包封率(d3),di和DF分别按以下公式计算。
粒径(d1)及PDI(d2)计算公式:di=(Ymax-Yi) / (Ymax-Ymin) ;包封率(d3)计算公式:di=(Yi-Ymin) / (Ymax-Ymin) ;综合评分 DF计算公式:DF=0.25×d1+0.25×d2+0.50×d3。其中,0.25,0.25及0.50分别为粒径、PDI和包封率的权值;Yi为各项测得值,Ymax、Ymin为各项测得值中最大值及最小值。
由表6,7可知,4个因素R值大小排列为A>B>C>D,表明泊那替尼用量、单硬脂酸甘油酯用量、蛋黄卵磷脂用量和泊洛沙姆188用量对P-SLN制备的影响程度逐渐减弱。各因素水平分析结果分别为A2>A1>A3、B1>B2>B3、C3>C2>C1、D3>D1>D2。其中,泊那替尼用量(A因素)对实验结果有统计学意义(P<0.05),其余因素差异无统计学意义。选取各因素的最高水平,并综合直观分析结果,初步确定最佳处方组合为A2B1C3D3,即泊那替尼用量10 mg,单硬脂酸甘油酯用量50 mg,蛋黄卵磷脂用量100 mg,泊洛沙姆188用量150 mg。
2.6形貌观察 按照优化后的处方、工艺制备P-SLN,用镊子夹着铜网沾取少量P-SLN混悬液,用2%磷钨酸染色2~3 min,并用滤纸吸取多余的溶液,自然晾干,置于透射电子显微镜下观察,并摄影(图2)。从图中可看出所制纳米粒为类球形粒子,大小分布比较均匀。

图2 P-SLN透射电镜照片(×25 000)
2.7粒径分布及Zeta电位测定 将所制P-SLN混悬液以纯化水稀释10倍,Zetasizer ZS90型激光粒度仪测定纳米粒子的粒径、粒径分布(图3)及Zeta电位(图4)。从图可看出纳米粒子粒径分布比较均匀,平均粒径为155.1 nm,PDI为0.152,Zeta电位为-22.0 mV。

图3 P-SLN粒径分布图
2.8验证性实验 按照优化后的处方工艺制备3批P-SLN,测定其粒径、电位及包封率,结果见表8。从表8可看出,采用乳化蒸发-低温固化法制备P-SLN,重复性良好。
2.9体外释药实验 精密移取按优化后的处方制备3批P-SLN混悬液1 mL,加入到最大通过相对分子质量(DA)为8000~10 000透析袋,除去透析袋中气泡,两端用线系好,置于100 mL含有1%聚山梨酯-80的磷酸盐缓冲液(pH值=7.4),置于恒温摇床,保持释药温度为(37±1)℃,转速100 r·min-1,分别于0.5,1.0,2.0,4.0,6.0,8.0,10.0,12.0,24.0 h等不同时间间隔取样1 mL,同时补充同温同体积释药介质1 mL。过膜,按含量测定方法测定含量,计算累积释放率,绘制累积释药曲线,实验结果见图5。另按处方量,制备泊那替尼与空白固体脂质纳米粒物理混合混悬液(P solution),按上述方法测定其体外释药累积曲线,实验结果见图5。
以释药方程拟合P-SLN混悬剂的累积释药曲线。由累积释药结果所得,拟合方程最佳的为:Weibull方程:lnln[1/(1-R)]=0.644 3lnt-1.044 3,r=0.986 9。

图4 P-SLN Zeta电位分布图

表8 P-SLN平均粒径、PDI、Zeta电位和包封率

图5 P-SLN的体外累积释放曲线(n=3)
2.10P-SLN冻干粉的制备 通过预实验,选择海藻糖和甘露醇作为P-SLN的冻干保护剂。取一系列不同浓度以及不同组分的冻干保护剂处方溶液,与P-SLN混悬液按1:1体积比例,各取2 mL,置于西林瓶中。在-74 ℃超低温冰箱中预冻24 h,取出在真空冷冻干燥机干燥24 h,即得P-SLN冻干粉。实验处方及结果见表9。综合考虑,P-SLN冻干粉外观、复溶时间、复溶后形态以及粒径变化,以5%甘露醇冻干保护剂的P-SLN冻干粉表现最为理想,冻干前后变化最小,确定以此处方制备P-SLN冻干粉。
2.11差动热分析仪(differential scanning calorimetry,DSC)物相分析 采用CDR-4P DSC进行P-SLN冻干粉样品物相分析。分别取适量泊那替尼原料药、辅料、处方量混合的物理混合物和P-SLN冻干粉,以空坩埚(Al2O3)为参比,10 ℃·min-1速度进行DSC扫描,实验结果见图6。
3 讨论
制备P-SLN的方法主要有薄膜分散法、高温乳化-低温固化法、高压均质法、微乳法等[18-22]。其中,高压均质法因其良好的可靠性和可重复性被广泛应用[23]。但高压均质对大规模生产提出了较高的设备要求,使用成本和能耗较大。在高温乳化-低温固化法中引入高剪切均质,可以明显缩短其制备过程,优化参数指标。其中,固化方法对制剂有明显影响,冰水浴结合高剪切固化法,可使粒径以及PDI明显减小。在高速均质中,乳化均质时间对制剂参数有较大影响,时间过短,乳化不充分,所制备的粒径和PDI较大,均质时间过长,可能破坏已经形成的稳定系统,造成粒径和PDI大幅改变。本研究表明,高速均质优化的高温乳化-低温固化法,简易可行,工艺可控,具有较好的可靠性和可重复性。
在脂质材料选择上,山嵛酸甘油脂、硬脂酸以及单硬脂酸甘油酯等脂质材料用于制备泊那替尼固体脂质纳米粒。山嵛酸甘油脂、硬脂酸制备的纳米粒在1周内发生沉降,纳米粒稳定性较差,单硬脂酸甘油酯制备的纳米粒表现良好,故最终确定脂质材料为单硬脂酸甘油酯。由于脂质材料在常温下为固态,制备温度应控制在高于脂质熔点5~10 ℃或10 ℃以上,使药物和脂质混合熔融。并且,乳化温度会影响乳化剂的乳化能力,温度过高会破坏乳化效果。单硬脂酸甘油酯的熔点在约55 ℃,在制备过程中温度应高于此值。通过实验,确定纳米粒制备的最佳乳化温度为(75±2)℃。
体外释药中,泊那替尼固体脂质纳米粒在最初的30 min内约10%主药突释,其中的原因为吸附在粒子表面的药物首先得到释放,随后是脂质核中的药物释放[24]。在研究中P-SLN释药前期,表面的药物扩散与P-solution的释放趋势相似。随着时间的推移,脂质纳米粒逐渐溶解,包封于纳米粒核心中药物被缓慢的释放,在24 h内累计释药70%[25-26]。同时,超过90%的主要从P-solution在仅12 h内释放出来,表明P-SLN除初期有较轻的突释效果外,体外缓释效果明显。
冷冻干燥可以用于提高SLN的稳定性,将液体SLN混悬液固化,可以降低纳米粒出现奥斯特瓦尔德熟化和水解反应,避免纳米粒在储存过程中发生粒子凝结和聚结,从而提高SLN稳定性。同时,将SLN转化成固体形式,可以将其作为药物中间体,进一步制备成片剂、丸剂或直接填充制备成为胶囊。此外,固化后,不需要特殊的储存条件和运输设备,极大地方便了药物的存储和运输。笔者使用5%甘露醇作为冷冻保护剂,制备的P-SLN冻干粉平均粒径前后变化最小,溶解时间最短,复溶后溶液更稳定。
表9 P-SLN冻干粉制备处方及实验结果

A.单硬脂酸甘油酯;B.卵磷脂;C.泊洛沙姆188;D.泊那替尼;E.P-SLN辅料物理混合物;F.P-SLN。
Fig.6DSCscanningimage
P-SLN的DSC物相分析结果显示,泊那替尼原料药在114.217 ℃有一个吸收峰,在136.808和141.155 ℃处有两个放热峰。辅料的DSC图谱显示,泊洛沙姆188唯一吸收峰在于57.936 ℃,单硬脂酸甘油酯的仅有吸收峰在62.986 ℃处,蛋黄卵磷脂未出现明显的吸收峰,可能与其为多种组分构成的混合物有关。P-SLN和P-SLN物理混合物的吸收峰位移,根据Gibbs-Thompson效应分析[27],两者的物相发生了转变,P-SLN不为辅料的物理混合物,而是一种全新的新物相,证明泊那替尼已经被成功包封于P-SLN的脂质核中,形成了新的物相组成形式[28]。
笔者在本实验中采用高速均质优化的高温乳化-低温固化法制备P-SLN新型制剂,方法简单可靠,质量可控,重复性好。同时,在与原料药的体外释药实验中,所制备P-SLN缓释效果显著,可以明显延长药物的释药时间,增加药物的生物利用度,为P-SLN的进一步研究提供了基础。
猜你喜欢
中国安全生产科学技术(2022年8期)2022-09-21
合成材料老化与应用(2021年6期)2021-12-23
上海理工大学学报(2021年3期)2021-07-20
中国蜂业(2019年3期)2019-04-03
中国兽医杂志(2018年9期)2018-12-29
中成药(2018年9期)2018-10-09
西南石油大学学报(自然科学版)(2018年1期)2018-02-10
中成药(2017年5期)2017-06-13
铁道科学与工程学报(2015年5期)2015-12-24
中国卫生标准管理(2015年32期)2015-07-18
